
記住我
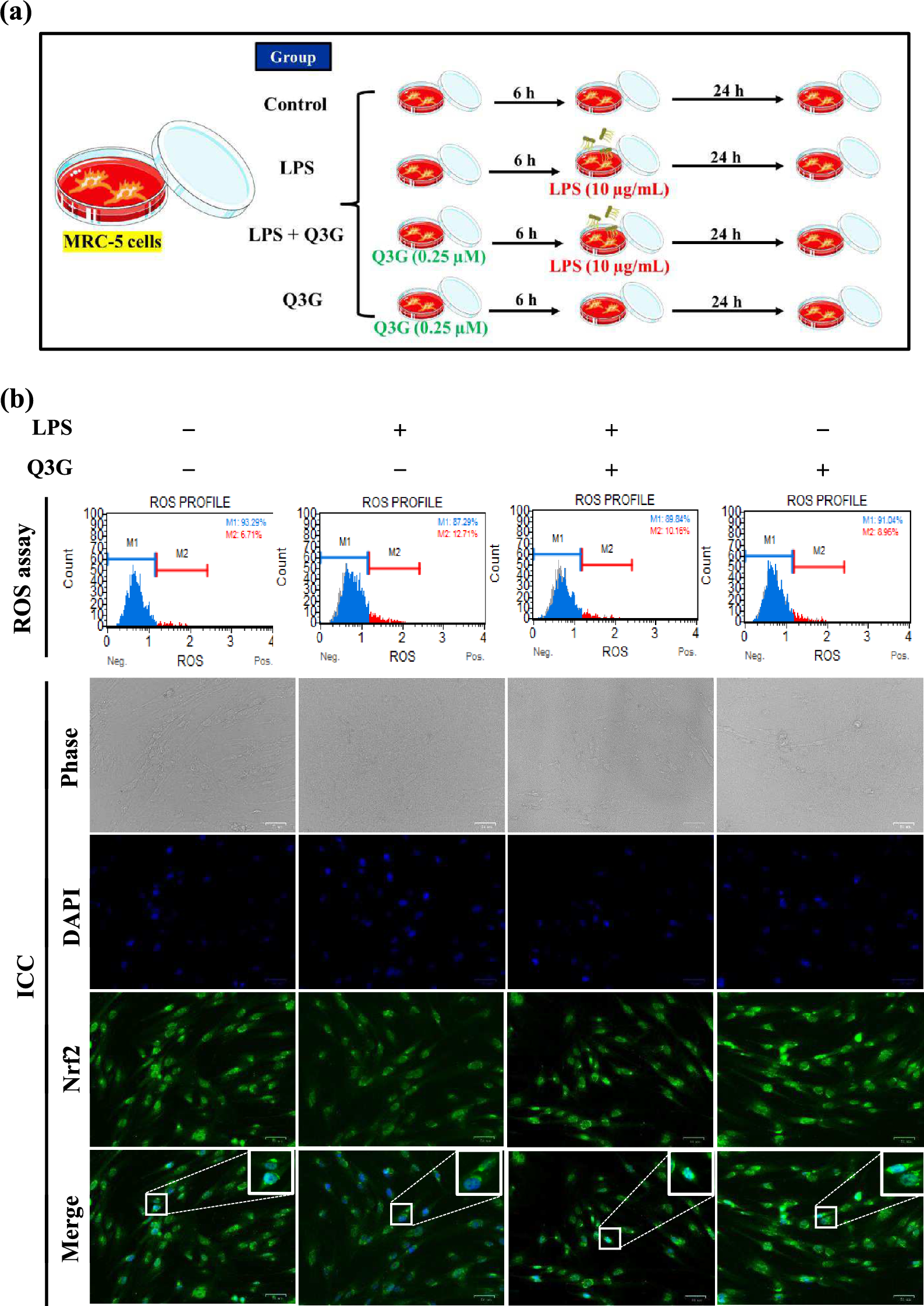
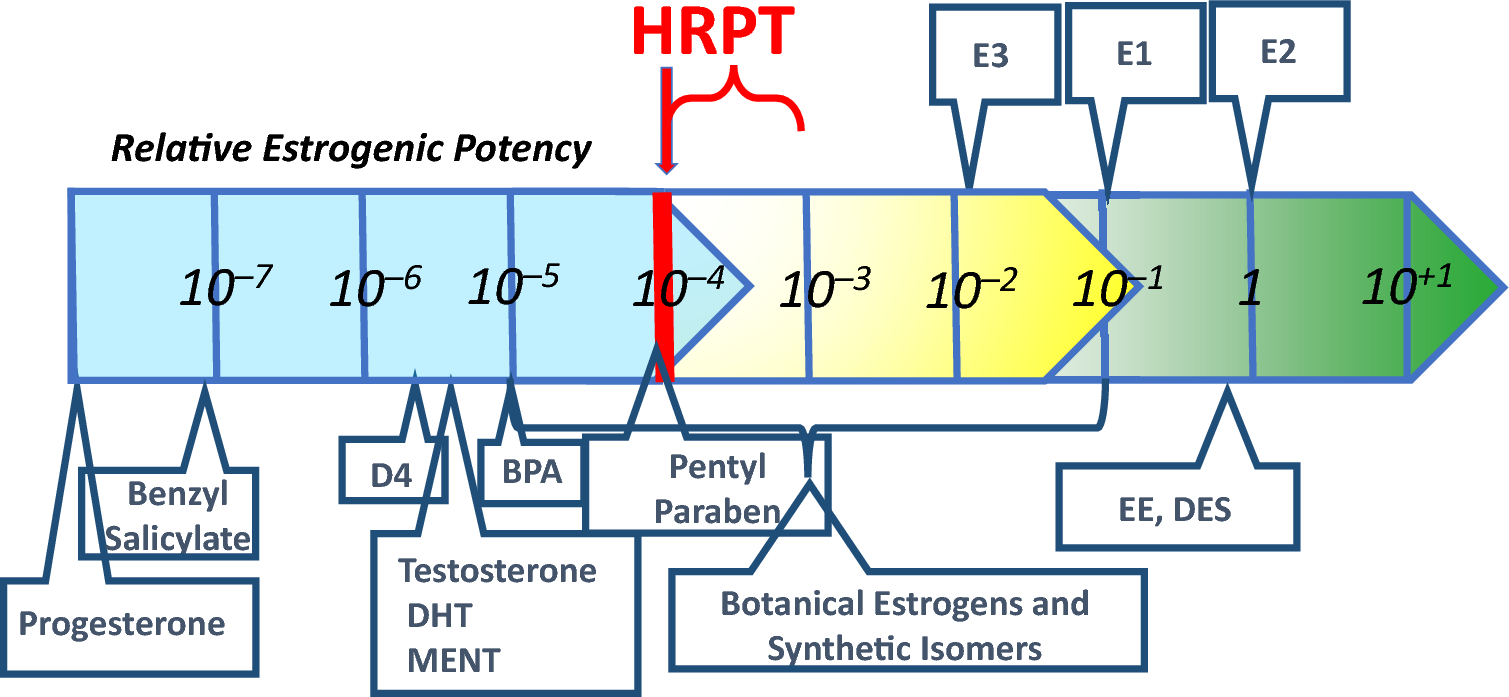
Table 2 lists the results of fractional receptor occupancy calculations for the primary endogenous estrogen 17β-estradiol (E2) alone and in the presence of five endogenous ERα ligands. As shown in Table 2, Row Q, E2 alone, in the absence of any competing ERα ligands, would occupy only about 7% of estrogen receptors at minimal plasma concentrations, similar to those that have been measured in pre-pubertal boys, but would nearly saturate estrogen receptors (~ 95%) at plasma concentrations similar to those observed in post-pubertal, non-pregnant women. It is important to appreciate that such calculations yield artificial, non-physiological representations of receptor occupancy because they consider only the endogenous primary hormone in the absence of competition by the endogenous metabolic milieu that is present naturally in the blood and bodily fluids. Similarly, most in vitro receptor binding and receptor transactivation assays used in toxicology to assess interaction with estrogen receptors do so in the absence of competing ligands, both exogenous (e.g., phenol red) and endogenous (e.g., components of serum). This is done to maximize sensitivity, but the results should be interpreted in light of the physiological-irrelevance of such conditions, as should receptor occupancy calculations that mimic them.
Table 2 Fractional receptor occupancy of sub-HRPT chemical in the presence of normal plasma concentrations of endogenous steroidal ERα ligandsTable 2, Row R shows the fraction of ERα occupied by E2 in the presence of five endogenous ERα-ligands, B–F, at mid-point and at minimal concentrations found in human blood. At minimal plasma concentrations of this subset of the endogenous metabolic milieu, the fractional receptor occupancy by E2 would be reduced only slightly—by about 1.4% (from 6.8381 to 5.4223%)—relative to the fraction occupied by E2 in the absence of this endogenous milieu (Row Q). This strongly suggests that a change in ERα-receptor occupancy of 1.4% is not relevant in humans, even at the most sensitive life-stage when E2 levels are lowest, and assuming that the endogenous metabolic milieu is also at its lowest. At mid-point plasma concentrations, the endogenous metabolic milieu is calculated to occupy a much greater fraction of ERα, and the fractional receptor occupancy by E2 would be reduced dramatically, from approximately 95% (94.8347%) for E2 alone (Row Q) to approximately 12% (11.733%) in the presence of the five additional endogenous ERα ligands (Row R).
Relevant to the hypotheses tested here, Row R shows that the normal range of ERα occupancy by E2 may be 5.4–11.7%. If one considers that a range of approximately 5.4–11.7% of ERα is occupied by E2 under normal physiological conditions, alterations of E2 receptor occupancy within this range may be considered unlikely to produce physiological effects and therefore, would be physiologically unable to alter endocrine function. A comparison of Rows R and Q shows the importance of considering the endogenous metabolic milieu when evaluating the potential for a chemical to produce physiologically relevant effects via a particular hormonal pathway. These calculations also indicate that the endogenous metabolic milieu is a primary determinant of ERα occupancy by the primary endogenous estrogen, E2. The endogenous milieu may be a more significant modulator of E2 activity during adult life stages when E2 levels are higher due to the concomitant higher concentrations of the other endogenous ERα ligands. Taken together, these calculations strongly suggest that to produce a physiologically relevant estrogenic effect, an alteration in E2 fractional ERα occupancy greater than 5% would be required.
The results of calculations shown in Table 2, Rows S and T are central to testing the hypotheses proposed herein. Table 2, Row S shows that when the receptor occupancy calculations include a hypothetical exogenous ERα ligand with affinity of 10–4 relative to E2, equal to the HRPT proposed previously (Borgert et al. 2018), the ERα occupancy due to E2 is essentially unaltered. This is regardless of whether the exogenous ligand is present at 1 nM amidst minimal plasma levels of the endogenous milieu (5.4223 to 5.4183% ≈ 0.004%) or at 1 µM amidst the mid-point plasma concentrations of the endogenous milieu (11.7533 to 11.6785% ≈ 0.075%). Row T shows that even if the exogenous ligand has affinity tenfold greater than the HRPT (10–3 relative to E2), no physiologically relevant change in receptor occupancy would be produced by this ligand at 1 µM amidst mid-point plasma levels of the five endogenous ligands (11.7533 to 11.0461% ≈ 0.7%), or at 1 nM amidst minimal endogenous ligand concentrations (5.4223 to 5.3826% ≈ 0.04%). These calculations provide compelling mechanistic support for the ERα-agonist HRPT proposed previously (Borgert et al. 2018) and the specific hypothesis tested here, based on the following considerations: (1) that exogenous ligands with affinity even tenfold greater than the HRPT (10–4 relative to E2) change E2 fractional receptor occupancy by less than 5%, a physiologically irrelevant change; (2) that the mid-point plasma concentration of the hypothetical exogenous ligand was assumed to be several orders of magnitude higher than is likely achievable for most exogenous chemicals (1 µM); and (3) that the minimal plasma concentration assumed (1 nM) for the exogenous ligand is also quite high, e.g., more than 200 times the maximum plasma concentration measured for one of the most well-studied exogenous chemicals, bisphenol A (BPA) (Mielke and Gundert-Remy 2009; Pande et al. 2019; Teeguarden and Hanson-Drury 2013; Teeguarden et al. 2013, 2016).
Calculations presented in Table 2, Rows U through Y allow a discussion of estrogenic activity in the broader context of total ERα occupancy by endogenous and hypothetical exogenous ligands and a more detailed consideration of the HRPT. Row U shows that the six endogenous ERα ligands considered here would occupy greater than 99% of ERα at normal concentrations measured in non-pregnant, post-pubertal women (“mid-point”), but only about 26% at the minimal plasma concentrations assumed to be present in pre-pubertal males. Row V shows that even amidst the minimal plasma concentrations of the endogenous milieu, a physiologically-relevant change in ERα occupancy would not be produced by introduction of an exogenous ERα ligand with affinity equal to HRPT potency (10–4 relative to E2), even if that ligand were present at a concentration of 1 nM (26.1809% versus 26.1263%). For comparison, 1 nM is more than 200-fold higher than the serum concentration measured for the most well-studied putative exogenous estrogen, BPA. Under those conditions, the exogenous ligand would contribute only 0.054% to the total ERα occupancy (Row X). Row W shows that no physiologically relevant change in ERα occupancy would be achieved even if the exogenous ligand had affinity tenfold higher than the HRPT potency amidst minimal concentrations of the endogenous milieu (26.6681% versus 26.1263%). Under those conditions, which assume a concentration of exogenous ligand likely unachievable in humans, the exogenous ligand would still contribute only 0.5417% (Row Y). Through similar comparisons, Rows V and W also show that amidst mid-point concentrations of the endogenous milieu, exogenous ligands with affinities equal to, or tenfold higher than the HRPT potency would produce no physiologically-relevant change in ERα occupancy when present at 1 µM, accounting for only 0.0041% and 0.0385% receptor occupancy, respectively.
Finally, the analysis was extended to ask for the potency or concentration at which an exogenous ligand would need to be present to overcome the potency threshold created by the endogenous metabolic milieu, even when present at only minimal concentrations. Row Z shows that to alter E2 receptor occupancy to a physiologically-relevant degree (> 5%), an exogenous ligand would need to have either an affinity approximately two orders of magnitude above the HRPT (10–2 relative to E2) if present in blood at 1 nM concentration, or, would need to achieve a concentration in blood of approximately 10 nM if its potency were only tenfold the HRPT (10–3 relative to E2).
It is important to appreciate that receptor occupancy calculations depend on both the potency and the circulating concentration of the ligand. Although it is sometimes assumed that any substance capable of interacting with a hormone receptor can produce an effect with sufficient exposure, this is clearly naïve. The receptor occupancy analysis shown here considered potential human exposures to exogenous substances but showed that the presence of a formidable endogenous metabolic milieu prevents the manifestation of estrogenic effects at concentrations physiologically achievable for most weak ERα ligands. This is evident from the earliest descriptions of reduced fertility in sheep. Although all sheep graze on plants that contain numerous botanical estrogens, reduced fertility occurred in those that grazed exclusively on a type of red clover that contains a high proportion of botanical estrogens with potencies above the HRPT of Borgert et al. (2018), such as Biochanin A, formononetic, coumestrol and genistein (Wyse et al. 2022). Thus, to be credible, hazard identification of EDCs should consider the potency of the substance at the molecular target and physiologically achievable concentrations of the substances (Autrup et al. 2015, 2020), and as demonstrated by the current analysis, whether physiologically achievable concentrations can overcome baseline occupancy of the molecular target by the endogenous metabolome.
The results shown in Table 2, Rows X, Y and Z have important implications for the ERα agonist HRPT originally proposed by Borgert et al. (2018). The results shown in Rows U–Z suggest that the HRPT estimate of 10–4 relative to E2 may be conservative by as much as tenfold, since ligands with affinities as high as 10–3 that of E2 would theoretically be unable to produce a physiologically relevant change in ERα occupancy, even when only five components of the endogenous metabolic milieu and the primary endogenous hormone, E2 are present. Figure 1 provides a revised version of Fig. 1 from Borgert et al. 2018, showing the potential range of conservatism in the threshold potency extending beyond the originally proposed HRPT to as high as 10–3 relative to E2. Figure 1 also adds potency estimates for three chemicals that have been incorrectly alleged to be exogenous estrogens. Potency data for benzyl salicylate and BPA (Natsch et al. 2021) and for parabens (Fayyaz et al. 2021) reveal that these chemicals lack the potency to disrupt the estrogen pathway as agonists or antagonists.
Fig. 1
Revised Human-Relevant Potency-Threshold (HRPT) for the ERα-Agonist MoA. This Figure was adapted from Borgert et al. (2018) showing the conservatism in the threshold region beyond the originally proposed HRPT to as high as 10–3 relative to E2, in accordance with receptor occupancy calculations shown in Table 2. Also added to the original figure by Borgert et al. (2018) are potency estimates for three chemicals incorrectly alleged to be exogenous estrogens. Potency data for benzyl salicylate and BPA are from Natsch et al. (2021) and data for parabens are from Fayyaz et al. (2021)
In addition to endogenous ERα ligands considered in the calculations shown here, the endogenous metabolome includes many other ligands that can interact with estrogen receptors, including ERα. For example, 27-hydroxycholesterol has been deemed a physiologically-active Selective Estrogen Response Modifier (SERM) in humans based on an IC50 value of 1 µM, an EC50 value of approximately 50 µM, and a plasma concentration 0.15–0.9 µM (DuSell et al. 2008; He and Nelson 2017). Androgens also bind estrogen receptors, albeit with low affinity. The presence of these additional endogenous chemicals that interact with the estrogen receptor strengthens the results and interpretations of receptor occupancy calculations shown here.
The analyses shown here explain three observations. First, they explain why extracts of low-affinity botanical estrogens failed to elicit measurable estrogenic effects in early clinical trials but showed weak clinical efficacy in more recent trials that used extracts standardized to a higher proportion of the most potent botanical estrogens (Borgert et al. 2018; Messina 2014). Borgert et al. (2018) relied on a comprehensive review of clinical trials with soy-based supplements (Messina 2014) that showed clinical outcome was dependent on the isoflavone profile of the product. Messina (2014) discussed studies showing that efficacy depended on meeting a threshold intake of genistein, and that an intake of at least 19 mg genistein per day could be predicted to show 60% efficacy in hot-flash reduction. Borgert et al. (2018) also relied on laboratory studies with soy extracts in which uterotrophic effects in Wistar rats were observable with administration of a 46% soy extract containing a high ratio of high-potency components (genistin, genistein and glycitein) compared to lower-potency components, but not with administration of an equal amount of a 51% soy extract that contained a low ratio of those high-potency components (de Lima Toccafondo Vieira et al. 2008). Thus, administration of low-potency isoflavones—those with potencies near the HRPT, such as daidzein—produced no uterotrophic effect, while administration of high-potency isoflavones—those with potencies above the HPRT, such as genistein—was uterotrophic. This indicates that efficacy is explained not primarily by exposure, but according to whether the extract contains components with sufficient potency to overcome the background receptor occupancy of the endogenous metabolic milieu.
The above explanation is consistent with our calculation that a functionally significant change in E2 receptor occupancy might be achieved by a ligand with relative potency of 1E-03 at a blood concentration of 1E-05 M (Table 1, Row Ib as shown by Table 2, Row Z). Results of several studies that reported on botanical estrogen levels in humans indicate that serum concentrations of botanical estrogens with potencies above the HRPT can, under some circumstances, reach concentrations that could alter the functional status of ER-mediated pathways. Although in healthy women age 25 years and older who were not taking medications or dietary supplements, mean genistein levels have been reported to be in the range of 25–300 nM (Arai et al. 2000; Mortensen et al. 2009; Palma-Duran et al. 2015), women enrolled in a cardiovascular survey, whose blood levels were validated against samples from women taking an isoflavone supplement in a clinical trial, had median serum genistein and equol levels in the range of 140 µM (median) – 340 µM (mean) (Barsky et al. 2021). Adult men taking an isoflavone formulation in a clinical trial reached mean serum levels about tenfold lower, in the range of 27 µM (Busby et al. 2002). Infants fed soy-based formulas have been reported to attain mean serum genistein concentrations in the low µM range (Cao et al. 2009; Mortensen et al. 2009; Ryowon et al. 2004; Setchell et al. 1997). Thus, humans can attain levels of genistein that might, in some individuals, produce clinically observable effects. As discussed above, this is consistent with results from more recent clinical trials of dietary supplements containing botanical estrogens, which show a low degree of efficacy (Messina 2014).
Second, the analysis of receptor occupancy shown here also explains why the male and female reproductive tract abnormalities caused by DES, including vaginal adenocarcinoma, exhibited a clear threshold (Borgert et al. 2012; Dietrich 2010; Golden et al. 1998; Hoover et al. 2011), even though during the era that DES was used during pregnancy, exposures to weakly estrogenic and anti-androgenic environmental contaminants, such as chlorinated pesticides, polychlorinated biphenyls and chlorinated dioxins were much higher than current exposures and blood levels (Dietrich 2010; Golden et al. 1998). As noted in the methodology section of this paper, circulating concentrations of estradiol peak during the follicular phase of the menstrual cycle in post-pubertal women and reach levels fifty times higher than the “mid-point” condition during pregnancy. In humans, the fetus is exposed to exogenous and endogenous substances via the maternal circulation. Because estrogen receptor occupancy by E2 would be highest during pregnancy due to approximately 50-fold higher circulating E2 concentrations compared to the “mid-point” condition we assessed, it should be obvious that an exogenous substance incapable of occupying a functionally significant fraction of receptors at the mid-point concentration would be even less able to affect receptor occupancy during pregnancy. This is corroborated by the tragic experience with DES, an ERα ligand with potency well above the HRPT that exerts agonist and antagonist effects in utero and produced reproductive tract abnormalities and cancer in offspring when high doses were administered to pregnant women during the first trimester of pregnancy. As explained previously (Borgert et al. 2012), if the endocrine system had already been “activated” above the threshold by endogenous estrogens, to which had been added exposure to environmental estrogenic EDCs, then any additional exposure to a strong estrogen such as DES should have produced observable effects. However, environmental exposures to ERα ligands with potencies below the HRPT were irrelevant. As shown here, this is explained by their inability to displace a functionally significant fraction of E2 and evidenced by the fact that only the highest-dose DES regimens produced reproductive abnormalities. As shown by receptor occupancy theory, instead of providing the basis for additive effects, the endogenous metabolic milieu provides a buffer of receptor occupancy that blunts the ability of weak ligands to alter the functional status of the system. Our quantitative explanations for those observations strengthen the conclusion that generalized additivity approaches are scientifically untenable for mixtures of putative endocrine disruptive chemicals (Borgert et al. 2012).
Third, the analysis presented here indicates that dismissing the role of potency in identifying chemicals with potential to elicit effects via endocrine mechanisms—i.e., potential endocrine disruptors—is based on a misunderstanding or mischaracterization of the components of potency. First, the receptor occupancy calculations shown here demonstrate that under physiological conditions, hormone receptors are in a state of continuous equilibrium binding due to the overwhelming concentrations of endogenous components of the metabolome that interact with hormone receptors. It is untenable to posit that a very small change in ligand binding can result in a physiologically-relevant change in receptor occupancy under non-equilibrium conditions because non-equilibrium conditions do not exist physiologically. Second, pharmacological potency is a function of both the affinity and efficacy of an interaction between a small molecule (e.g., receptor ligand) and a functional biological macromolecule (e.g., a receptor) (Borgert et al. 2013, 2018). Neither aspect can be dismissed. A ligand with affinity so low that it cannot substantially affect receptor occupancy cannot produce a physiological effect, irrespective of its efficacy at the receptor. This is mathematically obvious for receptor antagonists based on the calculations shown here for competition with the endogenous ligand 17β-estradiol.
Efficacy was not considered in the calculations shown here for the sake of simplicity, but a consideration of efficacy reinforces the HRPT concept and the physiological basis of thresholds for endocrine disruptive effects; efficacy works in concert with laws of mass action to dictate cellular and biochemical responses. Because of its low receptor occupancy, a low-affinity ligand is unlikely to produce a cellular response, even if the ligand possessed intrinsic activity equal to the primary endogenous hormone. Furthermore, efficacy is determined by the particular pattern of gene expression that each different ligand induces. This forms the fundamental basis of the selective estrogen response modifier (SERM) concept (Gronemeyer et al. 2004; Kuiper et al. 1999). Since each ligand may produce a unique conformational change in the receptor that produces a different cellular signal, simultaneous ER activation by different ligands—e.g., a weak partial agonist and a full agonist is unlikely to alter receptor signaling. Cellular responses typically require the simultaneous activation of several receptors per cell to elicit a cellular response (Borgert et al. 2013 and citations therein). Unless multiple receptors are activated simultaneously within a cell by ligands with the same or very similar intrinsic activity, cellular response is unlikely to be produced. Thus, although affinity considerations are sufficient to establish the molecular/biochemical basis of the HRPT concept, as shown here, it is further strengthened by an understanding of the dual role of efficacy with the laws of mass action.
Receptor occupancy calculations shown here demonstrate that low-potency ERα-ligands, defined as those having relative potencies at or below the HRPT for ERα-agonists, have an infinitesimally low probability of displacing a sufficient fraction of bound estradiol from estrogen receptors to elicit a physiological response or to alter the functional status of the system. This would hold not only for single chemicals but also for mixtures of low-potency chemicals. There are several reasons that mixtures of low-affinity ligands are likely to be incapable of altering the functional state of the estrogen pathway. Our receptor occupancy calculations indicate that such mixtures would displace less than 5% of bound estradiol from estrogen receptors, even under excessive exposure assumptions and importantly, when competition from the endogenous metabolic milieu is not ignored.
Thus, the work presented here also has important implications for the assumption that mixtures of exogenous chemicals act additively, conferring upon putative EDC mixtures the ability to be harmful even when concentrations of the individual chemicals are too low to cause effects on their own. It appears that those theories have been developed without consideration of the physiologically relevant mixture of chemicals that exist naturally. This includes not only hormones but also an overwhelming excess of endogenous metabolites with potency and/or affinity at hormone receptors greater than that of most putative EDCs. In light of the work presented here, it would seem prudent to reconsider the validity of such theories on mixtures, especially their recommended adoption for regulatory purposes by some governmental scientific bodies (NRC 2009).
The physiological and biochemical basis of the HRPT demonstrated here is corroborated by the work of Pande et al. (2019), who evaluated the individual and total estrogenic contribution of endogenous and exogenous estrogens measured in human serum. They developed a method that integrated approaches for measuring total hormone concentrations and calculated the bioavailability of hormones at concentrations found in serum. Similar to the approach taken herein, they used equations to resolve multiple equilibria between estrogenic ligands and receptors. They found that fractional receptor occupancy at ERα and ERβ was dominated by E1, E2 and E3, as was the total estrogenic response. This included ligand specific differences in recruitment of co-activator proteins (RCA), which further corroborates our assertion that the biochemical and physiological basis underlying potency thresholds extends beyond receptor-mediated mechanisms. Receptor occupancy by BPA—a chemical with potency below the HRPT—was at least five orders of magnitude lower than E1, E2 or E3, and three orders of magnitude lower than the fetal derived E4, genistein, or daidzein, contributing less than 1/1000th of the normal daily variability in total serum estrogenicity in a cohort of pregnant women.
留言 (0)