
記住我
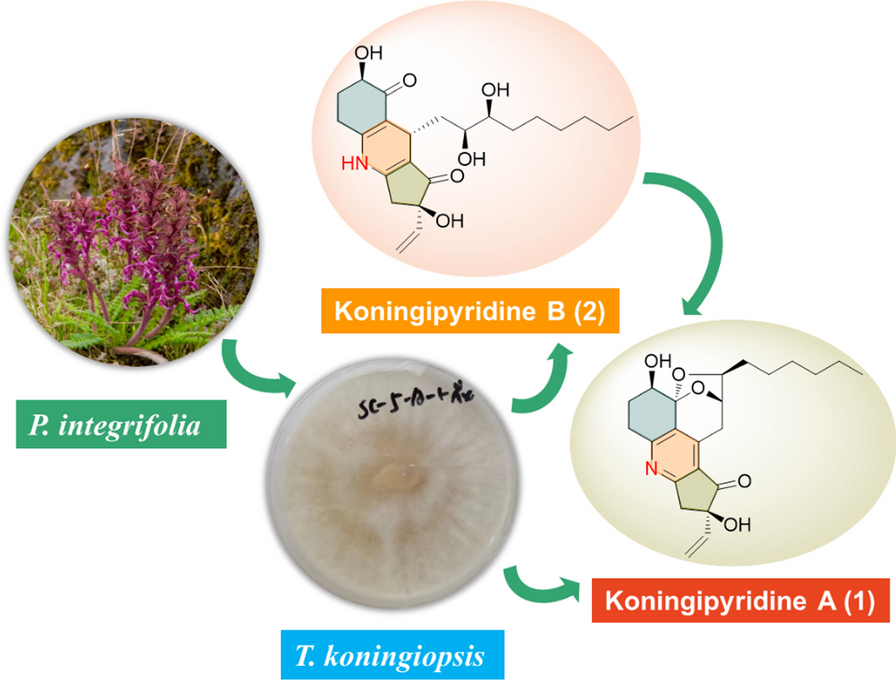
Koningipyridine A (1) was isolated as a yellow oil. It had the molecular formula of C23H29NO5 responsive for 10 degrees of unsaturation referring to the positive HRESIMS ion peak at m/z 400.2117 [M + H]+ (calcd for C23H30NO5+, 400.2118). The 1H NMR spectrum coupling with heteronuclear singular quantum correlations (HSQC) spectra of 1 showed a set of characteristic vinyl signals at δH 5.94 (1H, dd, J = 17.4, 10.8 Hz, H-22), 5.41 (1H, dd, J = 17.4, 0.6 Hz, H-23a), 5.23 (1H, dd, J = 10.8, 0.6 Hz, H-23b), three oxygenated methines at δH 4.57 (1H, dd, J = 4.8, 1.8 Hz, H-9), 4.08 (1H, dd, J = 4.2, 1.2 Hz, H-4), and 3.96 (1H, td, J = 7.2, 1.8 Hz, H-10), as well as a marginal methyl group at δH 0.89 (3H, t, J = 6.6 Hz, H-16). Moreover, the 13C NMR (Table 1) coupling with the DEPT 135 spectra disclosed 23 carbons consisting of one carbonyl group at δC 204.9 (C-18), nine methylenes at δC 28.2 (C-2), 26.8 (C-3), 33.5 (C-8), 36.1 (C-11), 26.5 (C-12), 30.5 (C-13), 33.1 (C-14), 23.8 (C-15), and 45.1 (C-20), three methines at δC 69.3 (C-4), 78.2 (C-9), and 83.1 (C-10), seven aromatic or olefinic carbons including a vinyl group at δC 116.0 (C-23) and 139.3 (C-22), together with five quaternary carbon atoms at δC 163.4 (C-1), 131.0 (C-6), 145.8 (C-7), 125.7 (C-17), and 170.8 (C-21), two oxygen-bearing quaternary carbons at δC 104.5 (C-5) and 81.1 (C-19), as well as one methyl group at δC 14.6 (C-16).
Table 1 1H (600 MHz) and 13C (150 MHz) NMR data of 1 and 2The aforementioned NMR features pointed to the conclusion that compound 1 should possess a complex pentacyclic fused ring system, which could be further constructed through the chemo-logical interpretation of 1H–1H COSY and HMBC spectra. The 1H–1H COSY cross peaks of H2-2/H2-3/H-4, H-22/H-23a/H-23b, and H2-8/H-9/H-10/H2-11/H2-12/H2-13/H2-14/H2-15/H3-16 of 1 suggested the existence of three independent fragments: a (C-2/C-3/C-4), b (C-22/C-23), together with c (C-8/C-9/C-10/C-11/C-12/C-13/C-14/C-15/C-16) as depicted in Fig. 2. On basis of the fragment a, the critical HMBC correlation signals from H2-2 to C-4/C-6, H2-3 to C-1/C-5, and H-4 to C-2/C-6 indicated the presence of cyclohexene ring A. Secondly, the obvious HMBC correlation signals from H2-8 to C-6/C-7/C-9/C-10, H-9 to C-5/C-7, as well as H-10 to C-5/C-8/C-9 and 1H–1H COSY cross-peaks of H-8/H-9/H-10 further constructed the rings D and E. Besides, as referring to the critical 1H–1H COSY fragment c, the side aliphatic chain was verified to be located at C-10 position of ring E with the aid of the key HMBC correlation signals from H-10 to C-12 and H-9 to C-11 (Fig. 2). The linkage for the three core rings A, D, and E was constructed as a 6/6/5 fused tricyclic pyran-ketal bridge skeleton, which was closely similar to the sub-structure of koninginin A [41] and koningiopisin C [21], two secondary metabolites also derived from the fungi of genus Trichoderma.
Fig. 2
Key 1H-1H COSY and HMBC correlations of compounds 1 and 2
In addition, the existence of an unprecedented cyclopentanone ring C in 1 could be confirmed through the obvious HMBC correlation signals from H-20 to C-17, C-18, C-19, and C-21. The terminal double bond was linked at the C-19 position of the C ring by the obvious HMBC correlation signals from H-22 to C-18 and C-20 as well as H-23 to C-19. Finally, by the comprehensive consideration of chemical formula, molecular unsaturation, together with NMR shifts for C-1 and C-21 of compound 1, the connection of rings A and C was deduced to be linked through C-1-N–C-21 bond with the construction of a penta-substituted pyridine ring B. Then, the planar architecture of 1 had been completely constructed to feature an unprecedented 6/6/5/6/5 fused pentacyclic skeleton with a natural rarely-occurring penta-substituted pyridine ring system, and it was exampled as the first member with a nitrogen-containing skeleton reported in the koninginin family.
The relative configuration of 1 was tentatively determined with aid of the NOESY spectrum. In the NOESY spectrum, the cross peak between H-4 and H-9 revealed that the protons of H-4 and H-9 ought to be assigned on the same side as α-orientation, whereas the C-5/O/C-9 should be on the other side (Fig. 3). Moreover, the cross peaks between H-4 and H-11, H-12 were also disclosed, and it strongly suggested that the side chain (C-10 to C-16) should be β-orientation, when H-4 is α-orientation. However, the determination of the relative configuration of C-19 terminal vinyl moiety was seemed to be an intractable challenge, because it was far away from the chiral centers in rings A, D, and E, which thus resulted in the lack of the NOESY correlation.
Fig. 3
Key 1H-1H NOESY correlations of compound 1
In order to solve this bleak problem of the relative configuration at C-19, the 13C NMR calculation was applied. The GIAO-DFT 13C NMR calculations [42, 43] for the two probable candidate diastereoisomeric structures 1a and 1b were performed at the ωB97x-D/6-31G* (IEFPCM, CD3OD) level [44, 45]. As a result, both of the correlation coefficient (R2) values for 1a and 1b were 0.9991 (Fig. 4), these results indicated that the previous inference of the planar structure and partial relative configuration (C-4/C-5/C-9/C-10) of compound 1 could be significantly trusted. Moreover, the resulting Prel value of 1b was 90.48%, while that of 1a was only 9.52% (Table 2), which collectively pointed to that the diastereoisomeric 1b or its enantiomer is more likely to be the correct relative structure for 1.
Fig. 4
Regression analyses of experimental and calculated 13C NMR chemical shifts for 1a and 1b
Table 2 Calculated 13C chemical shifts (CD3OD) of structures 1a and 1b fitting to the experimental data of 1With the hope to verify the relative configuration of compound 1 and determine its absolute configuration, the TDDFT-ECD theoretical calculations in Gaussian16 involving the diastereoisomers 1a and 1b together with their corresponding enantiomers were applied. Experimentally, geometric optimizations of the probable isomers of 1a and 1b were conducted to discover the desired conformers with minimum energy, and the TDDFT methodology was then implied at the ωB97x-D/TZVP theory level. Furthermore, the related conformers were Boltzmann averaged to get calculated ECD spectra of 1a and 1b. As the results showed in Fig. 5, the experimental ECD curve of 1 had been disclosed to be much more consistent with the calculated one of 1b by comparing the experimental and calculated ECD curves. Notably, this result was also perfectly matched with the conclusion of the 13C NMR DFT calculations. Therefore, as referring to the aforementioned reliable results, the absolute configuration of 1 was finally deduced to be 4R,5S,9S,10S,19R and named koningipyridine A.
Fig. 5
Experimental and calculated ECD spectra of compound 1 (in MeOH)
Koningipyridine B (2) was purified as a yellow oil. Its chemical formula C23H33NO6 was determined on the basis of the positive HRESIMS ion peaks at m/z 420.2394 [M + H]+ (calcd for C23H34NO6+, 420.2386) and 442.2224 [M + Na]+ (calcd for C23H33NO6Na+, 442.2206), which indicated the existence of eight degrees of unsaturation in 2. A further careful inspection and comprehensive elucidation of the NMR spectra for 2 revealed its chemical structure with a certain similarity to that of 1. Moreover, the 1D NMR spectral data also showed a set of vinyl group signals at δH 5.76 (1H, dd, J = 17.4, 10.8 Hz, H-22), 5.28 (1H, dd, J = 17.4, 1.2 Hz, H-23a), 5.08 (1H, dd, J = 10.8, 1.2 Hz, H-23b) as well as δC 114.0 (C-23) and 140.4 (C-22) together with four non-proton olefinic carbons at δC 153.9 (C-1), 111.5 (C-6), 115.1 (C-17), and 162.5 (C-21).
Besides, two keto-carbonyl functional groups at δC 201.0 (C-18) and 198.5 (C-5) were observed. The aforementioned established functional groups accounted for five degrees of unsaturation, which suggested that compound 2 ought to be the ketal ring-opening product of 1 with a tricyclic system by the consideration of the existence of the two hydroxyl groups in the side aliphatic chain and a keto-carbonyl moiety in the cyclohexene ring A.
The planar structure of 2 could be further evidenced by analysis of its 1H–1H COSY and HMBC spectra. There were three independent spin fragments from the 1H–1H COSY spectrum: a (H2-2/H2-3/H-4), b (H-22/H2-23), and c (H-7/H2-8/H-9/H-10/H2-11/H2-12/H2-13/H2-14/H2-15/H3-16) as shown in Fig. 2. A close comparison of the 1H–1H COSY correlations of 2 with those of compound 1 could readily find the same fragments a and b. Moreover, the HMBC correlations in the rings A–C of 2 were also almost the same as those of 1. The aforementioned results indicated that compound 2 might share a similar 6/6/5 skeleton (rings A–C) as that of 1.
However, the chemical shift of C-5 at δC 104.5 in 1 was downshifted to δC 198.5 in 2, it suggested that the ketal functionality in 1 was replaced by a carboxyl unit in 2. In addition, the non-proton olefinic carbons in 2 were one less than those in 1, which might be attributed to the change of olefinic C-7 carbon (δC 145.8) in 1 to a methine one (δC 25.3) in 2. All these aforementioned facts indicated that compound 2 was a D/E ring-opening derivative of 1. Notably, the HMBC correlations from H-7 to C-5/C-9, H2-8 to C-6/C-17 and a lack of HMBC correlations from H-9 and H-10 to C-5 confirmed the aforementioned conclusion. Therefore, the planar structure of 2 was established (Fig. 2), and it was tentatively suggested to be a critical biosynthetic precursor for 1.
Similarly, we tried to determine the relative configuration of 2 by analyzing its NOESY spectrum, whereas there were not any obvious diagnostic NOESY correlations being observed. Thus, the determination of the relative configuration of compound 2 through the NOESY experiment seemed to be bleak. Therefore, an extensive combination of chemical transformation, Mo2(OAc)4-induced electronic circular dichroism (Mo-ICD) spectrum, GIAO DFT 13C NMR calculations, and theoretical ECD calculation were applied to solve the stereochemistry of compound 2.
Firstly, with the consideration of the presence of 9,10-vicinal diol in 2 from a biogenetic synthetic perspective, the absolute configuration of C-9/C-10 of 2 was tentatively deduced to be the same as that of compound 1. In order to provide much more direct evidence, the chemical transformation of the 9,10-vicinal diol with acetone to generate the five-membered ketal ring was tried to determine the relative configuration. The treatment of compound 2 with acetone in the presence of a catalytic amount of PTSA (p-toluenesulfonic acid) obtained its acetonylidene derivative (Fig. 6), which possessed a trans-acetonylidene configuration on the basis of the critical NOESY correlations of H-9/H3-24 and H-10/H3-25 (Fig. 7A). Therefore, compound 2 was revealed to be a threo-9,10-diol.
Fig. 6
Chemical transformation of 2
Fig. 7
Configuration determination of the 9,10-diol in 2
Then, Snatzke and Frelek’s methodology was applied to establish its absolute configuration with the aid of the Mo-ICD spectrum [46,47,48,49,50,51,52,53]. As the results compiled in Fig. 7B, the obvious positive Cotton effects at 338 and 400 nm originated from the positive (Mo–O)-C–C-(O–Mo) torsion angle. The careful analyses of the locally favored conformations for the two isomers of the threo-9,10-diol, it could be readily found that the 9S,10S-isomer featured an unambiguously positive torsion angle, whereas the 9R,10R-isomer owned a negative one (Fig. 7C). Therefore, the 9S,10S configuration of 2 was assigned.
Although the absolute configuration of 9,10-vicinal diol was fortunately determined by chemical transformation and Mo-ICD methodology, the remaining three unassigned chiral centers still led to eight possible related diastereoisomers. In order to further confirm the relative configuration of 2, the GIAO-DFT 13C NMR calculations towards four candidate structures 2a–2d were also attempted. Unfortunately, the relative configuration of 2 could not be completely addressed by 13C NMR calculations due to the Prel values of 2a (48.20%) and 2d (43.36%) as shown in Table 3. Moreover, the R2 values for the linear regression of 13C NMR data were revealed to be 0.9991 for 2a and 0.999 for 2d (Fig. 8), which strongly indicated that both 2a and 2d or their enantiomers might be the correct structure for 2.
Table 3 Calculated 13C chemical shifts (DMSO-d6) of structures 2a–2d fitting to the experimental data of Fig. 8
Regression analyses of experimental and calculated 13C NMR chemical shifts for 2a-2d
Subsequently, theoretical ECD calculations for 2a-2d were further applied to determine the absolute configuration of 2, it is delightful to find that the theoretical ECD spectrum of 4R,7S,9S,10S,19R-2a was well matched with the experimental one (Fig. 9), and both curves showed obvious positive cotton effects at 230, 260, and 330 nm together with a series of negative cotton effects at 210, 250, 300, and 350 nm. Moreover, the theoretical ECD calculations also supported the two potential probable structures 2a and 2d deduced by the GIAO DFT 13C NMR calculations. Collectively, the aforementioned experimental results thus disclosed the absolute structure of 2 as 4R,7S,9S,10S,19R.
Fig. 9
Experimental and calculated ECD spectra of compound 2 (in MeOH)
Interestingly, the extensively deduced absolute configuration of 2 was consistent with that of its derivative 1. From the perspective of biosynthesis, the consistent configuration of compounds 1 and 2 further proved the reliability of the aforementioned results. Therefore, compound 2 was identified as the first member in the koninginin family sharing a unique 6/6/5 dihydropyridine skeleton with an absolute configuration of 4R,7S,9S,10S,19R, and it was given the trivial name koningipyridine B.
Moreover, four koninginin derivatives were also isolated from the endophytic fungus T. koningiopsis SC-5 and identified as koninginin D (3) [29], koninginin E (4) [30], koninginin F (5) [31], and 7-O-methylkoninginin D (6) [3] by a careful comparison of their spectral data with the published data.
The novel scaffold of 1 was evidenced to feature an unprecedented pentacyclic ketal skeleton with the formation of a fascinating 6/6/5/6/5 fused ring system and shared a characteristic pyridine core, which represents the first example of nitrogen-containing koninginin type natural product. While, koningipyridine B was revealed to be the first member in koninginin family sharing a unique 6/6/5 dihydropyridine skeleton, and it was probably a critical biosynthetic precursor of koningipyridine A as proposed in the plausible biosynthetic pathway shown in Scheme 1.
Scheme 1
Plausible biosynthetic pathway of 1 and 2
Subsequently, all isolates were tested for antibacterial activities towards S. aureus, MRSA (Methicillin-resistant S. aureus), and E. coli by the microbroth dilution method [54], whereas they did not exhibit any notable antibacterial effect at the concentration of 100 μg/mL. In addition, compounds 1˗6 had been screened for cytotoxic activity towards three human cancer cell lines (A549, Hela, and HepG2) by MTT assay [55] as shown in Additional File 1: Table S1, and all of them showed weak activities.
留言 (0)