
記住我
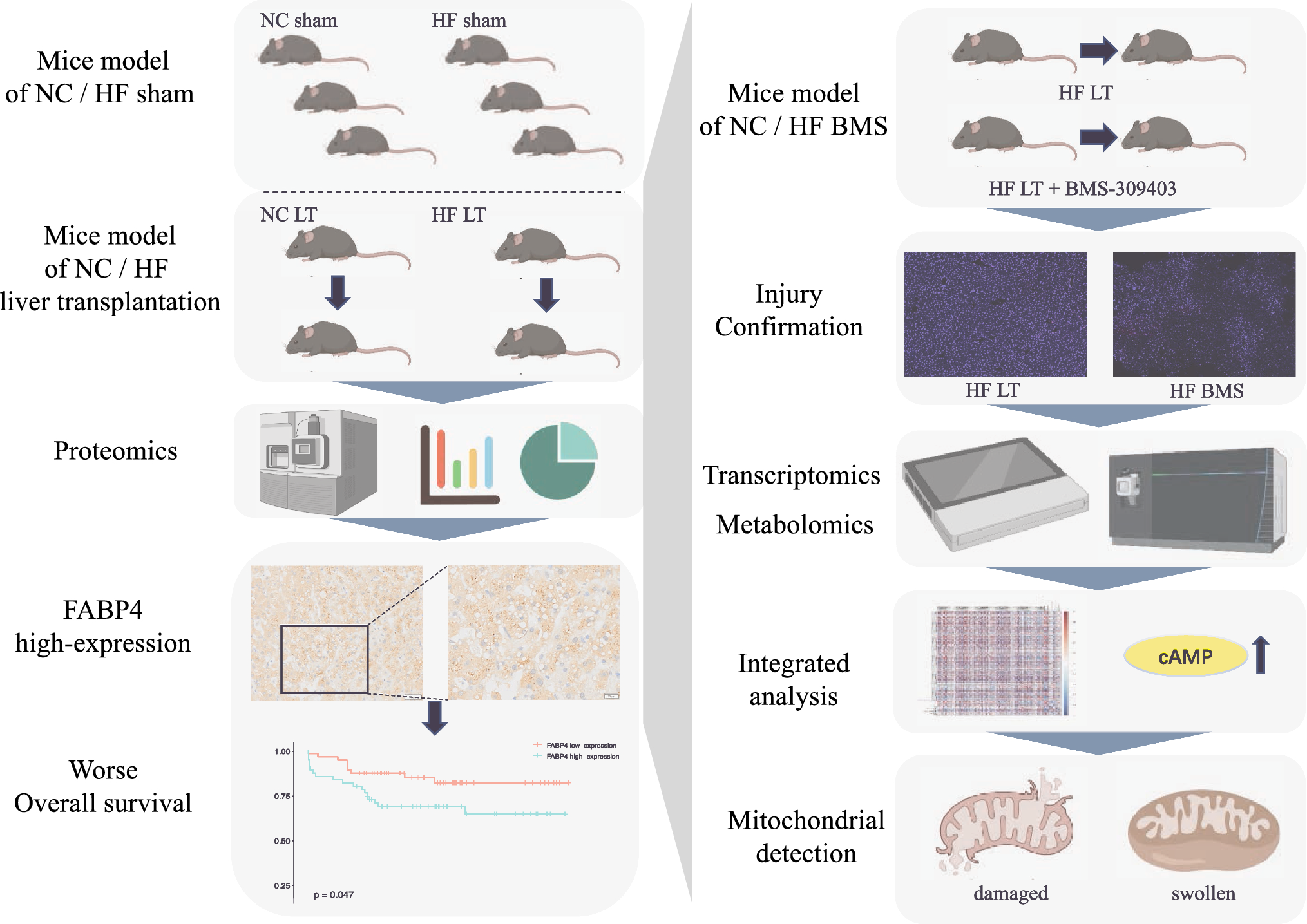
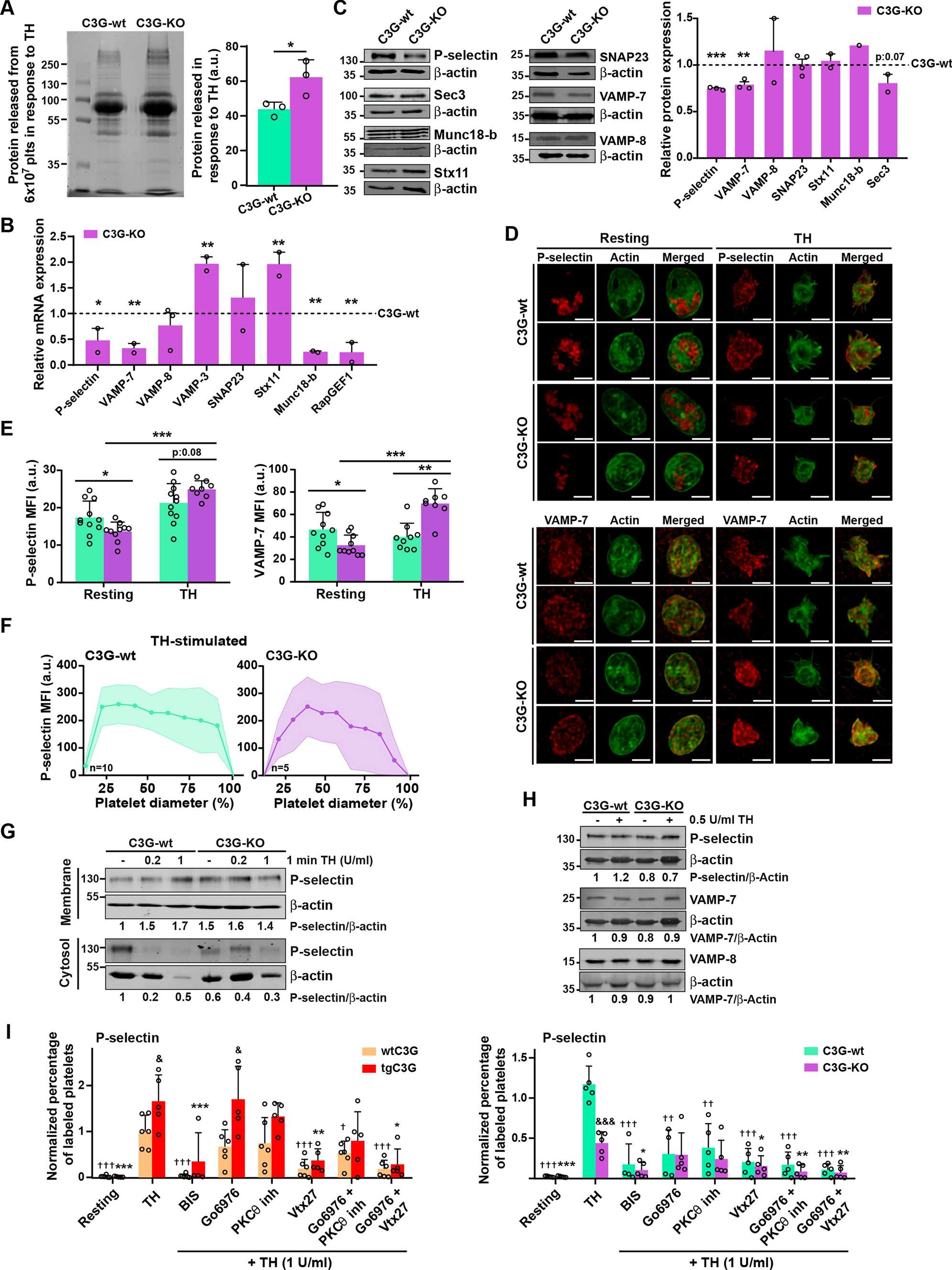
We previously described that the transgenic expression of C3G in platelets induces the retention of some angiogenic factors (e. g. VEGF, bFGF and TSP-1) through a mechanism independent of its GEF activity [7], suggesting that C3G could participate in α-granule secretion. Consistently, ablation of C3G in platelets results in increased secretion of proangiogenic factors [6]. In agreement with these results, we show here that the secretome of thrombin-stimulated C3G-KO platelets contained a higher net amount of protein than that of C3G-wt platelets (Fig. 1A). C3G ablation did not produce substantial changes in platelet size or structure, nor did it affect the number of α- or δ-granules (Fig. S2A-C). Therefore, the observed increase in platelet secretion points to a faulty secretory mechanism. Supporting this, we found in C3G-KO platelets altered expression of some of the components of the secretion machinery: lower expression of P-selectin, VAMP-7 and Munc18-b, and higher expression of VAMP-3 and Stx11 mRNAs (Fig. 1B and Table S1). A decrease in P-selectin and VAMP-7 was also detected at the protein level (Fig. 1C) and by immunofluorescence in resting platelets (Fig. 1D, E). These findings align with a specific role of C3G in platelet secretion. Unexpectedly, we observed a positive correlation between C3G and RasGRP2 expression in MKs, potentially explaining why RasGRP2 is unable to compensate for the C3G defect (Fig. S1D).
Fig. 1
C3G controls α-granule secretion through PKCδ. A SDS-PAGE of secretome proteins from C3G-wt and C3G-KO 6 × 107 platelets (plts) stimulated with 0.2 U/ml thrombin (TH) for 5 min at 1100 rpm and 37 °C. Proteins were visualized with Colloidal Blue Staining. Histogram represents the mean ± SD of the quantification of proteins in the releasate. B RT-qPCR analysis of the expression of the indicated genes in mRNA from C3G-KO and C3G-wt platelets using β-actin as housekeeping gene. C3G (RapGEF1) was used as control. C Western blot analysis of P-selectin, Sec3, Munc18-b, Syntaxin 11 (Stx11), SNAP23, VAMP-7 and VAMP-8 protein levels in lysates from C3G-wt and C3G-KO resting platelets. Bar graphs represent densitometric analysis of protein bands, relative to β-actin expression and normalized against wild-type platelets. D Representative immunofluorescence confocal microscopy images of C3G-wt and C3G-KO platelets stimulated with 0.5 U/ml thrombin (TH) for 1 min and labeled with antibodies anti-P-selectin + Cy3 (upper) or anti-VAMP-7 (H-55) + AF568 (lower) (red), and with phalloidin-iFluor 488 (green). Scale bar: 2 µm. E Bar graphs represent the mean ± SD of the MFI of P-selectin (left), or VAMP-7 (right) in resting platelets and in response to thrombin. F Line/scatter plots represent the mean ± SD of P-selectin distribution along the platelet diameter (MFI). G C3G absence alters P-selectin distribution in platelets. Western blot analysis of P-selectin levels in the membrane (upper) or cytosolic (lower) fractions from resting, 0.2 or 1 U/ml thrombin (TH)-stimulated C3G-wt and C3G-KO platelets. β-actin was used as loading control. Values of the P-selectin/α-actin ratio were normalized against those of resting C3G-wt platelets. H Western blot analysis of P-selectin, VAMP-7 and VAMP-8 levels in resting and 0.5 U/ml thrombin-stimulated C3G-wt and C3G-KO platelets. β-actin was used as loading control. Values are relative to β-actin expression and were normalized against resting C3G-wt platelets. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. I Washed blood from (left) wtC3G and tgC3G or (right) C3G-wt and C3G-KO mice was pre-treated with 1 µM Go6976 (an inhibitor of PKCα and β), 20 µM PKCθ inhibitor or 100 µM Vtx27 (wtC3G and tgC3G) or 2 µM Go6976, 40 µM PKCθ inhibitor or 200 µM Vtx27 (C3G-wt and C3G-KO) for 30 min, or with 5 µM BIS for 5 min, prior to stimulation with 1 U/ml thrombin (TH). Then, samples were incubated with anti-CD62P-FITC + anti-CD41-APC antibodies. Bar graphs represent the mean ± SEM of the percentage of P-selectin-FITC-labeled platelets normalized against TH-stimulated control platelets. & ≤ 0.05, &&& ≤ 0.001 versus their corresponding wild-type; †p ≤ 0.05, ††p ≤ 0.01, †††p ≤ 0.001 versus TH-stimulated wild-type; *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 versus TH-stimulated tgC3G (left) or C3G-KO (right). MFI Mean Fluorescence Intensity, a.u. arbitrary units
C3G-KO platelets exhibit reduced levels of Rap1-GTP, integrin αIIbβ3 activation, platelet aggregation and P-selectin exposure on the surface in response to various agonists [4] (Fig. S1E–H). Lower levels of P-selectin were also detected in the membrane of thrombin-activated C3G-KO platelets by immunofluorescence, with only 50% of platelets exhibiting peripheral distribution versus 70% of C3G-wt platelets (Fig. 1D, F). In fact, P-selectin was retained in the cytoplasm of these platelets after stimulation with 0.2 U/ml thrombin (Fig. 1G). In addition, C3G deletion altered the distribution of VAMP-7, but not VAMP-8, α-granules in resting platelets. Indeed, we observed two different distribution patterns of VAMP-7: a normal central distribution (C3G-KO-1) and a peripheral/membrane distribution (C3G-KO-2) (Fig. S2D). Furthermore, we found an increase in the MFI of P-selectin and VAMP-7 in C3G-KO platelets after thrombin stimulation (Fig. 1D, E), which was not due to an increase in protein levels (Fig. 1H). Therefore, the increased MFI could be due to granule fusion.
In contrast to the role of C3G in regulating α-granule exocytosis, we did not observe any effect of C3G deletion in lysosome (Fig. S2E, F) or δ-granule secretion (Fig. S2G, H). Similarly, neither overexpression of C3G or C3GΔCat mutant, nor C3G ablation had any effect on platelet endocytosis (Fig. S2I).
Overall, these results indicate that C3G would play a role in platelet α-granule exocytosis, a process regulated by PKC [20]. Specifically, PKCδ activates thrombin-dependent degranulation [41]. On the other hand, C3G participates in the thrombin-PKC pathway leading to Rap1 activation in platelets, and is phosphorylated in a PKC-dependent manner [3, 4]. We observed that PKCδ inhibition (with Vtx27, an inhibitor of PKCδ and PKCθ isoforms), but not PKCθ or classical PKC inhibitors, abrogated the increased P-selectin exposure induced by C3G transgene expression, with a minor effect on C3G-KO platelets, suggesting that PKCδ is involved in the mechanism by which C3G regulates α-granule exocytosis (Fig. 1I). Consistently, PKCδ was also involved in the activation of integrin αIIbβ3 by C3G (Fig. S2J).
C3G modulates the formation of Arp2/3-VARP-VAMP-7 and trans-SNARE complexes and participates in vesicle docking via Rap1-RalAWe previously showed that C3G interacts with VAMP-7 protein, mainly with its SNARE domain, in resting and thrombin-stimulated platelets [7]. Consistent with that, and with the above results indicating a putative role for C3G in platelet secretion, we found that platelet C3G also interacted with secretory machinery proteins VARP and VAMP-8 and also weakly with SNAP23, Stx11, and Munc18-b (Fig. 2A). No interaction with VARP and Munc-18b was detected in tgC3GΔCat platelets, suggesting the involvement of the C3G GEF domain in the formation of these complexes (Fig. 2B). Likewise, no interaction of C3G with components of the exocyst complex, such as Sec3, Sec5, Sec10, or Exo70 was observed (Fig. 2A). Some of these interactions were confirmed in co-transfected HEK293T cells (Fig. S3A).
Fig. 2
C3G interacts with proteins involved in vesicle trafficking and regulates Ral1 activation and the trans-SNARE and Arp-VARP-VAMP-7 complexes. A tgC3G or B tgC3GΔCat platelets, both at rest and after thrombin (TH) stimulation (0.5 U/ml for 1 min), were immunoprecipitated with anti-C3G G-4 antibodies and the levels of P-selectin, VARP, Sec3, Sec5, Munc18-b (tgC3G and tgC3GΔCat), Sec10, Exo70, CrkL, Stx11, SNAP23 and VAMP-8 (tgC3G) were detected by western blot. CrkL was used as a positive control. IP: immunoprecipitation; Stx11: Syntaxin 11. The SNAP23 panel in A is displayed as a composite panel. The raw data source file for this panel is illustrated in Fig. S5A. C Lysates from (left) wtC3G and tgC3G or (right) C3G-wt and C3G-KO platelets, both at rest and after thrombin (TH) stimulation (0.2 or 1 U/ml for 1 min) were immunoprecipitated with an anti-Arp2 antibody and the levels of VARP, VAMP-7 and VAMP-8 were detected by western blot. β-actin was used as loading control. IP: immunoprecipitation. Values are relative to corresponding total protein levels and were normalized against unstimulated wild-type platelets. Mouse IgG was used as a negative control. Green asterisks indicate non-specific bands (IgG). The red arrow points to VAMP-7 band. D Representative immunofluorescence confocal microscopy images of C3G-wt and C3G-KO platelets stimulated with 1 U/ml thrombin (TH) for 1 min and labeled with anti-VAMP-7 (H-55) + AF568 (red) and anti-Arp2 + AF647 (green). Scale bar: 2 µm. Bar graphs show the Pearson’s Correlation Coefficients (mean ± SD) of the intensity values of VAMP-7 and Arp2 under the indicated experimental conditions. E Lysates from (left) wtC3G and tgC3G platelets or (right) C3G-wt and C3G-KO platelets, both at rest and after thrombin (TH) stimulation (0.2 or 1 U/ml for 1 min) were immunoprecipitated with an anti-SNAP23 antibody and the levels of VAMP-7 and VAMP-8 detected by western blot. β-actin was used as loading control. IP: immunoprecipitation. Values are relative to levels in total lysate and are normalized to those of wild-type platelets. Mouse IgG was used as a negative control. F Representative immunofluorescence confocal microscopy images of C3G-wt and C3G-KO platelets stimulated with 0.2 U/ml thrombin (TH) for 1 min at rest or followed by 30 min of spreading, and labeled with anti-VAMP-7 (158.2) + AF647 (red) and anti-SNAP23 + AF568 (green). Scale bar: 2 µm. Enlarged images of a single platelet per genotype are depicted for a clearer visualization of colocalization. Arrowheads point to yellow pixels. Bar graphs show the Pearson’s Correlation Coefficients (mean ± SD) of VAMP-7 and SNAP23. G Pull-down assays to detect RalA activation after stimulation of tgC3G, tgC3GΔCat, C3G-KO platelets and their respective wild-types with 0.2 or 1 U/ml thrombin (TH) for 1 min. The levels of RalA-GTP were detected by immunoblotting with anti-RalA antibodies. Representative blots from 2–3 experiments are depicted. The line/scatter plots show the mean ± SD of RalA-GTP levels. Values are relative to total RalA levels. *p ≤ 0.05
VARP forms a complex with Arp2 and VAMP-7 during platelet α-granule secretion, in which VARP prevents unscheduled actin polymerization, required for α-granule fusion and platelet spreading [12]. C3G overexpression in platelets favored the Arp2-VARP interaction, while a decrease in the interaction of Arp2 with VARP, VAMP-7 and VAMP-8 was found in C3G-KO platelets (Fig. 2C). In addition, significantly lower colocalization was also observed between VAMP-7 and Arp2 in resting and thrombin-stimulated C3G-KO platelets (Fig. 2D). These results point to a negative regulatory role of C3G in platelet degranulation.
The interaction with SNARE proteins suggests that C3G could participate in the formation of the trans-SNARE complex. Indeed, a stronger interaction between VAMP-7 and SNAP23 was found in thrombin-stimulated C3G-KO platelets, both in suspension and spreading conditions (Fig. 2E, F), whereas C3G overexpression resulted in decreased SNAP23/VAMP-7 complex formation (Fig. 2E). Similarly, interaction between VAMP-7 and Stx11 increased in spread C3G-KO platelets (Fig. S3B). Consistently, the absence of SNAP23 increased C3G interaction with VAMP-7 and Stx11, with no effect on the C3G-VARP interaction (Fig. S3C–F). These results indicate that C3G would compete with SNAP23 for binding to VAMP-7 and Stx11, and explain its lower interaction with SNAP23 and Stx11 in tgC3G platelets after stimulation (Fig. 2A). Therefore, C3G would act as a negative regulator of trans-SNARE complex formation.
Hence, our results suggest that C3G could act as a brake on platelet α-granule secretion by reinforcing the VARP-VAMP-7-Arp2 complex, thus preventing the SNAP23-VAMP-7 interaction and, consequently, the trans-SNARE complex formation. This would explain the increased secretion in C3G-KO platelets after thrombin stimulation (Fig. 1A).
RalA/B GTPases, whose activity is regulated by Rap1, are involved in targeting vesicles to the PM during platelet exocytosis [14]. Thus, we next studied whether C3G actions in exocytosis could be mediated by RalA/B activity. Figure 2G shows that overexpression of C3G significantly increased RalA-GTP levels after stimulation with 1 U/ml thrombin, whereas overexpression of the C3GΔCat mutant or C3G deletion showed the opposite tendency. This result indicates that C3G participates in Rap1-mediated RalA activation in platelets. Therefore, the defects in exocytosis observed in tgC3G and C3G-KO platelets could be explained by defects in docking/tethering (controlled by RalA activation) and also alterations in downstream steps, such as vesicle priming and fusion, regulated by the formation of the trans-SNARE complex.
C3G promotes platelet spreading in a PKC- and substrate-dependent mannerOne of the consequences of platelet degranulation is platelet spreading, which is dependent on VAMP-7 α-granules [12, 16]. We previously showed that transgenic expression of C3G increased platelet spreading in a Rap1-independent manner [7]. Consistently, C3G-KO platelets showed a marked delay in spreading on ibiTreat plates, as measured by decreased platelet area (Fig. 3A and Videos 1 and 2). To gain insight into this process, we evaluated whether the role of C3G in platelet spreading was substrate-dependent. In line with the above results, deletion of C3G in platelets resulted in defective spreading of thrombin-stimulated (0.2 U/ml) platelets on poly-l-lysine, type I collagen, fibronectin and vitronectin (Fig. 3B). In concordance, and in agreement with previous results [7], tgC3G platelets showed the opposite behavior on the same substrates (Fig. 3C). In contrast, C3G expression did not affect spreading on fibrinogen or laminin (Fig. 3B, C). These results indicate that the role of C3G in spreading depends on the substrate. In addition, tgC3GΔCat platelets exhibited increased spreading on poly-l-lysine and type I collagen, but decreased spreading on vitronectin, with no defects on fibronectin (Fig. S4A). This intermediate phenotype between that of tgC3G and C3G-KO platelets indicates that C3G would regulate platelet spreading through GEF-dependent and -independent mechanisms. Similar results were obtained with high dose thrombin (1 U/ml, data not shown).
Fig. 3
C3G promotes platelet spreading on poly-L-lysine, collagen, fibronectin and vitronectin through PKC. A Representative images of C3G-wt and C3G-KO platelets spread on ibiTreat plates at different times (0, 30, 60, 120, 300 and 480 s). Bar graphs represent the mean ± SD of platelet area, measured with ImageJ software. Scale bar: 5 µm. B, C Representative images of B C3G-wt and C3G-KO platelets or C tgC3G and wtC3G platelets resting or spread on poly-L-lysine, type I collagen, fibrinogen, fibronectin, laminin and vitronectin for 30 min after 0.2 U/ml thrombin stimulation. Platelets were stained with phalloidin-iFluor 488 to visualize actin cytoskeleton. Scale bar: 5 µm. Bar graphs represent the mean ± SD of platelet area (µm2). Substrates in which there are differences between genotypes are highlighted in red. D RT-qPCR analysis of the indicated integrin subunits in platelets from C3G-KO and C3G-wt mice using β-actin as housekeeping gene. Values were normalized to those of C3G-wt platelets. The expression of RapGEF1 (C3G) was used as control. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. E Representative images of wtC3G, tgC3G, C3G-wt and C3G-KO platelets spread on poly-L-lysine for 30 min after pre-treatment or not with 5 µM BIS for 5 min, followed by 5 min stimulation with 2 µM PMA. Platelets were stained with phalloidin-iFluor 488 to visualize actin cytoskeleton. Scale bar: 5 µm. Bar graphs represent the mean ± SD of platelet area (µm2). & ≤ 0.05, && ≤ 0.01, versus their corresponding wild-types; ††p ≤ 0.01, †††p ≤ 0.001 versus PMA-stimulated wild-type platelets; **p ≤ 0.01, ***p ≤ 0.001 versus PMA-stimulated tgC3G or C3G-KO platelets
The aforementioned results suggest that C3G could regulate the expression, or surface exposure, of integrins involved in the interaction of platelets with type I collagen, fibronectin or vitronectin. Thus, we analyzed in C3G-KO and control platelets the expression of subunits of the main platelet integrins, such as α2 (ITGA2), αIIb (ITGA2B), α6 (ITGA6), αV (ITGAV), αL (ITGAL), β1 (ITGB1), β2 (ITGB2) and β3 (ITGB3) by RT-qPCR [3, 4] using two different set of primers for each integrin (Table S1). We found significantly lower expression of the integrin subunits α6, αV and β1 in C3G-KO platelets than in C3G-wt platelets (Fig. 3D), which could explain the lower interaction of C3G-KO platelets on fibronectin, type I collagen and vitronectin [42]. However, we did not find alterations in the surface expression of GPVI (collagen receptor), integrins αIIb (ITGA2B or CD41) and β3 (ITGB3 or CD61), which bind to fibrinogen, fibronectin and vitronectin, and glycoprotein Ib alpha (GP1BA or CD42b), which binds to collagen, in tgC3G, tgC3GΔCat and C3G-KO platelets (Fig. S4B). This result suggests that C3G might be regulating the outside-in signaling triggered by collagen, fibronectin or laminin, probably affecting actin polymerization.
We next analyzed whether this C3G function was also dependent on PKC, given its involvement in platelet spreading [20]. As shown in Fig. 3E, the pan-PKC inhibitor bisindolylmaleimide (BIS) abrogated the differences in spreading on poly-l-lysine between the different genotypes after stimulation with PMA, indicating the involvement of PKC in the role of C3G in platelet spreading.
C3G regulates actin polymerization during lamellipodia formation via Rac1, but independently of Rap1Platelets spread by forming filopodia and lamellipodia, which involves profound changes in the actin cytoskeleton [43]. The in vivo spreading experiment in Fig. 3A, suggested defective lamellipodia formation in C3G-KO platelets. Based on that, we studied whether the abnormal spreading found in C3G-KO and tgC3G platelets were due to defective formation of these actin structures. To do so, we performed a time-lapse spreading assay, on different substrates, on thrombin-stimulated platelets, analyzing the formation of filopodia and lamellipodia after 5, 15, and 30 min of spreading. C3G-KO platelets were able to form filopodia on all substrates, but showed delayed lamellipodia formation on type I collagen, fibronectin, and vitronectin (Fig. 4A and Fig. S4C), the same substrates on which defective spreading was observed. Consistently, C3G overexpression triggered faster lamellipodia formation on type I collagen and vitronectin, although with smaller differences (Fig. 4B and S4D).
Fig. 4
C3G interacts with proteins involved in actin remodeling and regulates lamellipodia formation through Rac1 in a Rap1-independent manner. A, B Quantification of the different spreading phases in fixed platelets from A C3G-wt and C3G-KO or B wtC3G and tgC3G mice, after 30 min spreading on type I collagen, fibrinogen, fibronectin, laminin or vitronectin coated coverslips, induced by 0.2 U/ml thrombin for 1 min. Bar graphs represent the mean ± SEM of the percentage of platelets in the different spreading phases: I: round platelets; II: platelets with filopodia; III: intermediate state with both filopodia and lamellipodia; IV: spread platelets. wt: wild-type; tg: transgenic; KO: knockout. Asterisks refer to quantification of platelets in phase IV. Substrates showing differences between genotypes are highlighted in red. C, D Platelets from wtC3G and tgC3G or C3G-wt and C3G-KO mice were stimulated with 0.2 U/ml thrombin (TH) for 1 min and allowed to spread on CRP-XL (CRP) or fibrinogen (Fb) for 30 min before lysis (C), or C3G-wt and C3G-KO platelets in suspension were pre-treated with 10 µM latrunculin (LatA) for 45 min and stimulated with 0.2 U/ml thrombin (TH) for 1 min before lysis (D). The supernatant containing the soluble G-actin and the urea-solubilized triton X-100-insoluble pellet containing F-actin were subjected to SDS-PAGE and the levels of F-actin and G-actin were detected with anti-β-actin antibodies. Levels of β-actin from total lysates are also depicted. The numbers indicate the ratio F/G actin normalized to wild-type, non-treated platelets. Fb: Fibrinogen. E Washed platelets in suspension from (left) wtC3G and tgC3G or (right) C3G-wt and C3G-KO mice were stimulated with 0.2 or 1 U/ml thrombin (TH) for 1 min before fixing with 2% PFA. After permeabilization with 0.2% Triton X-100, platelets were stained with phalloidin-iFluor 488 and analyzed by flow cytometry. Bar graphs represent the mean ± SD of phalloidin-iFluor 488 MFI, normalized against resting control platelets. F Lysates from (upper) tgC3G and (lower) tgC3GΔCat platelets, resting or stimulated with thrombin (TH, 0.5 U/ml for 1 min) were immunoprecipitated with anti-C3G antibodies (anti-C3G G-4 in the case of WAVE1, Cofilin, Arp3; anti-C3G C-19 in the case of Abi1, β-Actin, WAVE2, Arp2 and CrkL) and the levels of WAVE1, WAVE2, Abi1, Cofilin, N-WASP, β-actin, VASP, Arp2, Arp3 and CrkL were detected by western blot. CrkL was used as a positive control. Red arrows indicate WAVE2 and Abi1 bands. The yellow asterisk indicates a non-specific band. IP: immunoprecipitation. The Arp2 and β-actin western blots and IPs are shown as composite panels. The raw data source file for these panels is depicted in Fig. S5B. G Pull-down assay to detect Rac1 activation after stimulation of tgC3G, tgC3GΔCat, C3G-KO platelets and their respective wild-types with 0.2 or 1 U/ml thrombin (TH) for 1 min. The levels of Rac1-GTP were determined using anti-Rac1 antibodies. Representative blots from 2 experiments are depicted. The line/scatter plots show the mean ± SD of Rac1-GTP levels. Values are relative to total Rac1 levels. *p ≤ 0.05, ***p ≤ 0.001. H Representative images of wtC3G, tgC3G, C3G-wt and C3G-KO platelets spread on CRP-XL (CRP) or fibrinogen for 30 min after pre-treatment with 50 µM 1A-166 or 50 µM CK-166 for 30 min, prior stimulation with 0.2 U/ml thrombin (TH) for 1 min. Platelet were stained with phalloidin-iFluor 488 to visualize actin cytoskeleton. Scale bar: 5 µm. Bar graphs represent the mean ± SD of platelet area (µm2) of (upper) wtC3G and tgC3G or (lower) C3G-wt and C3G-KO platelets. & ≤ 0.05 versus its wild-type. ††p ≤ 0.01, †††p ≤ 0.001 versus TH-stimulated wild-type spread platelets; **p ≤ 0.01, ***p ≤ 0.001 versus TH-stimulated tgC3G or C3G-KO spread platelets. MFI Mean Fluorescence Intensity, a.u. arbitrary units
The above results suggest a role for C3G in actin remodeling during the outside-in signaling. Therefore, we monitored actin fiber formation in tgC3G, C3G-KO platelets and their wild-types spread on CRP-XL and fibrinogen for 30 min, by analyzing the F-actin/G-actin ratio by western blot. As shown in Fig. 4C, C3G overexpression induced a slight increase in actin polymerization after spreading on CRP-XL, but not on fibrinogen. Consistently, and most evidently, C3G-KO platelets showed a decrease in F-actin levels after spreading on CRP-XL (Fig. 4C) or in suspension (Fig. 4D, E), but not on fibrinogen (Fig. 4C). All these results support a regulatory role for C3G in platelet cytoskeleton remodeling during spreading on type I collagen, but not on fibrinogen, consistent with the results in Fig. 3B–D. All these results suggest the involvement of C3G in outside-in signaling triggered by type I collagen, fibronectin and vitronectin, leading to actin polymerization.
To further characterize this role of C3G in spreading and since Src kinases regulate C3G phosphorylation [4] and platelet spreading [44], we analyzed spreading in the presence of the Src inhibitor PP2. PP2 abolished the increase in platelet area in tgC3G platelets, suggesting the involvement of Src in this C3G function (Fig. S4E). However, PP2 did not inhibit spreading in control platelets at the concentration and time used. Alternative pathways regulating platelet spreading in the absence of Src have been described [44]. Furthermore, we analyzed platelet spreading in tgC3G, C3G-KO and control platelets in the presence of reagents that interfere with actin polymerization, such as cytochalasin D (CytD) or latrunculin A (LatA). As shown in Fig. S4E, and as expected, CytD-and LatA-treated tgC3G and wild-type platelets were unable to spread. In contrast, C3G-KO platelets were affected by CytD, but not LatA treatment, which rather reverted the effect of C3G knockout. This suggests that C3G deletion would prevent the effect of LatA, which binds actin monomers and enhances F-actin depolymerization from both ends [45]. These data are reinforced by the observation that LatA reverted the decrease in F-actin/G-actin ratio induced by C3G deletion (Fig. 4D).
Given this new role of C3G in regulating actin cytoskeletal structures in platelets, we next examined whether C3G interacts with proteins involved in cytoskeletal remodeling. Immunoprecipitation assays showed that C3G interacted with WAVE2, Abi1, VASP, Arp2, and Arp3, but not with WAVE1, Cofilin, and N-WASP in resting and thrombin-stimulated platelets (Fig. 4F). CrkL and β-actin were monitored as positive controls of C3G interactions. Similar results were obtained in co-transfected HEK293T cells (Fig. S4F). These results suggest that C3G could regulate Rac1 activity in platelets. Indeed, we found higher levels of Rac1-GTP in thrombin-stimulated tgC3G platelets, compared to control platelets, while C3G-KO platelets showed the opposite trend (Fig. 4G). Surprisingly, overexpression of the C3GΔCat mutant also promoted Rac1 activation, suggesting that C3G would regulate Rac1 activity independently of Rap1 (Fig. 4G). No effects of C3G on RhoA or Cdc42 activation were detected (data not shown). To corroborate these findings, we monitored platelet spread on CRP-XL and fibrinogen in thrombin-stimulated tgC3G, C3G-KO platelets, and their controls in the presence of 1A-166 or CK-666, inhibitors of Rac1 and Arp2/3 complex, respectively. As expected, both inhibitors significantly reduced spreading on both substrates in all genotypes. However, tgC3G platelets spread on CRP-XL (but not on fibrinogen) were more sensitive to Rac1 and Arp2/3 inhibition than wtC3G platelets (Fig. 4H). This result supports the involvement of C3G in the Arp2-VARP-VAMP-7 complex (Fig. 2C), and further suggests that C3G would regulate Rac1 function during platelet spreading.
C3G deletion in platelets promotes kiss-and-run and compound exocytosisSo far, our results indicate that C3G would favor platelet spreading by promoting the Rac1-WAVE2-Arp2/3 pathway, while it would decrease platelet secretion by interfering with the trans-SNARE complex. The opposite was found in C3G-KO platelets, i.e., increased platelet secretion and decreased spreading. The C3G-KO platelet phenotype is consistent with a kiss-and-run exocytosis, similar to that seen in RalA/B double KO platelets [13]. To verify this, we monitored the spreading of C3G-KO and C3G-wt platelets on ibiTreat plates (Ibidi) after staining with FM1-43. While C3G-wt platelets showed decreased fluorescence over time, indicating a normal, fuse and collapse, exocytosis, two different exocytosis patterns were found in C3G-KO platelets: (i) fuse and collapse exocytosis (C3G-KO-1), with kinetics similar to that of C3G-wt platelets, (ii) kiss-and-run exocytosis (C3G-KO-2), in which FM1-43 signal is not lost, indicating no fusion of the granules with the PM (Fig. 5A-C). In addition, we observed a higher number of fused granules in C3G-KO platelets, compared to C3G-wt platelets, indicative of compound exocytosis (Fig. 5D), in agreement with results in Fig. 1D, E.
Fig. 5
C3G-KO platelets show kiss-and-run and compound exocytosis. C3G-wt and C3G-KO platelets were allowed to spread on a µ-Slide 8-well plate (Ibidi) after staining with 10 µm FM1-43. A Representative images of granules, taken every 10 s for 1 min. B Scatter plot representing the quantification of MFI (mean ± SEM) of individual FM1-43-stained granules from all C3G-wt and C3G-KO platelets analyzed. C Same analysis as in B but C3G-KO platelets were separated into two categories depending on their phenotype: fuse and collapse (C3G-KO-1) or kiss-and-run (C3G-KO-2) exocytosis. ***p ≤ 0.001. †††p ≤ 0.001 C3G-KO-2 versus C3G-wt. D Representative images showing single and compound granules. Bar graphs represent the mean ± SD of the percentage of single or compound vesicles in platelets of each genotype
C3G regulates granule exocytosis in PC12 cells, promoting protein releaseTo further demonstrate the involvement of C3G in granule secretion, we investigated whether C3G modulates the kinetics of dense-core vesicle exocytosis in NPY-td-Orange- expressing PC12 clones stably transfected with wild-type C3G (C3G-wt), a hyperactive C3G mutant (C3G-Y554H) [34], or a shC3G construct (pLVTHM-C3Gi) to silence C3G expression [5] (Fig. 6A). We used TIRF Microscopy to analyze the dynamics of vesicle-PM fusion events, by monitoring the kinetics of the tdOrange signal after KCl stimulation, a stimulus of granule exocytosis in these cells. Overexpression of wild type C3G resulted in a slightly lower AUC of tdOrange fluorescence, indicating faster secretion kinetics. In contrast, overexpression of C3G-Y554H mutant significantly slowed the kinetics of tdOrange-NPY release, manifested by increased AUC (Fig. 6B, C and Videos 2–5). On the other hand, C3G-silencing significantly increased the rate of tdOrange-NPY release (Fig.
留言 (0)