
記住我
The complement system, a key component of innate immunity, provides the first line of defense against microbial invasion. It also participates in the pathogenesis of many chronic, non-infectious human disorders, including autoimmune diseases, hyperacute graft rejection, paroxysmal nocturnal hemoglobinuria (PNH), atypical hemolytic uremic syndrome (aHUS), and atherosclerosis [1, 2]. Since its first identification in 1895 [3], our understanding of the complement system has led to the development of successful complement-targeted therapeutics used to treat diseases ranging from PNH/aHUS to the SARS-CoV-2 infection (COVID-19) [4]. It is well established that bacterial invasion activates the complement system, forming activated bioproducts and the terminal membrane attack complex (MAC), a terminal complement activation product, thereby culminating in lysis and pathogen clearance [5]. However, it is less clear how viral infections such as SARS-CoV-2 activate the complement system. Like in SARS-CoV-2, the complement system appears to be maladaptive in respiratory syncytial virus (RSV) infection, which can similarly cause acute respiratory distress syndrome (ARDS), a life-threatening lung injury that allows fluid to leak into the lungs [6, 7]. However, in other respiratory viral infections that can cause ARDS, such as influenza A & B, the role of the complement system is complicated. C3 may be protective while C3a and C5a signaling and MAC may be detrimental to the host [8, 9]. The cellular and molecular mechanisms by which the components of the complement system contribute to the pathogenesis of these respiratory viral infections remain unclear. We will review (1) complement activation and regulation, (2) clinical evidence of the detrimental role of the complement in COVID-19, (3) beneficial effect of complement-related therapeutics on COVID-19, 4) the pathogenic roles of the complement in other respiratory viral infections including influenza and RSV, and 5) experimental approaches to dissect the complement-mediated mechanisms during acute respiratory viral infection.
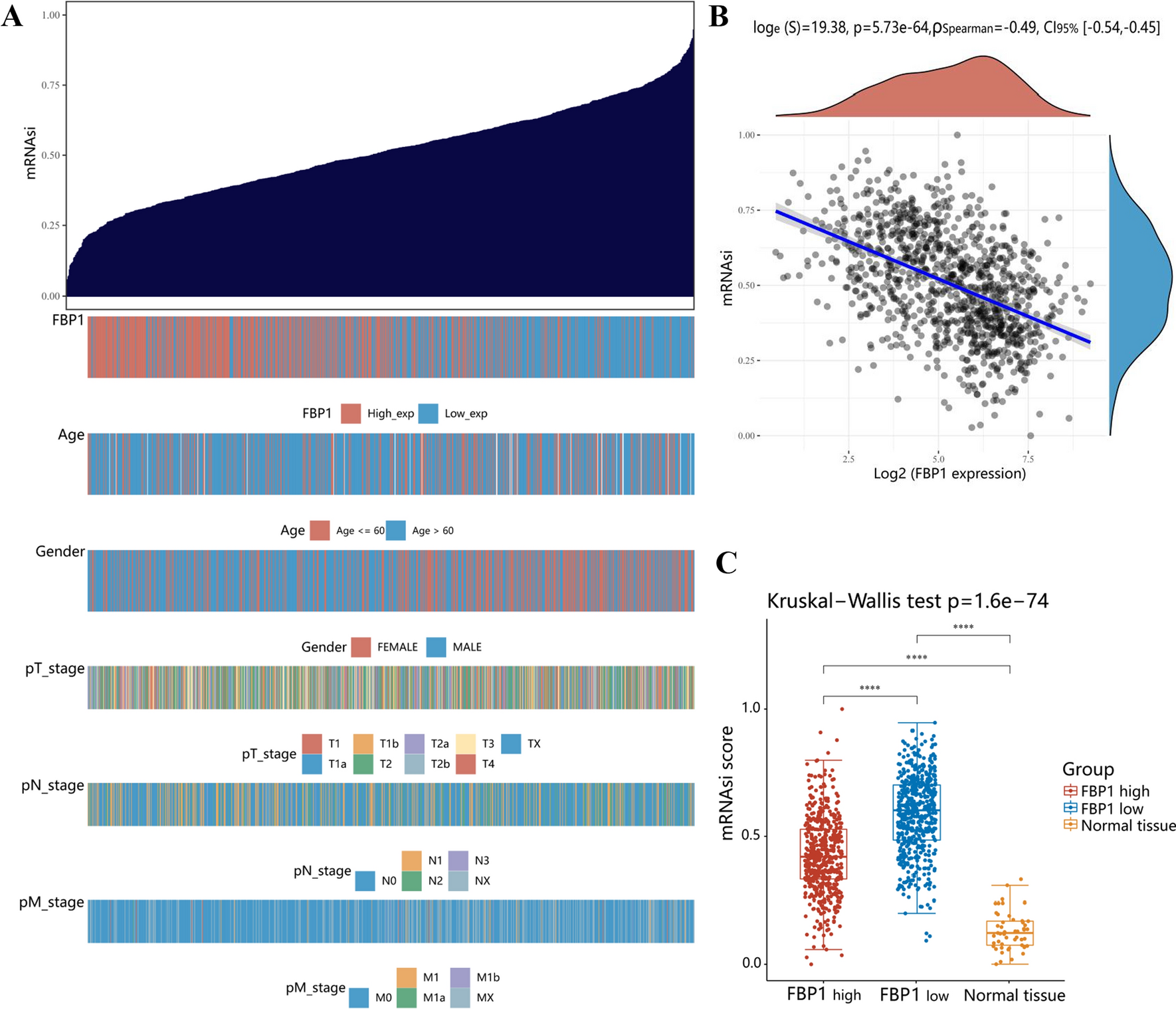
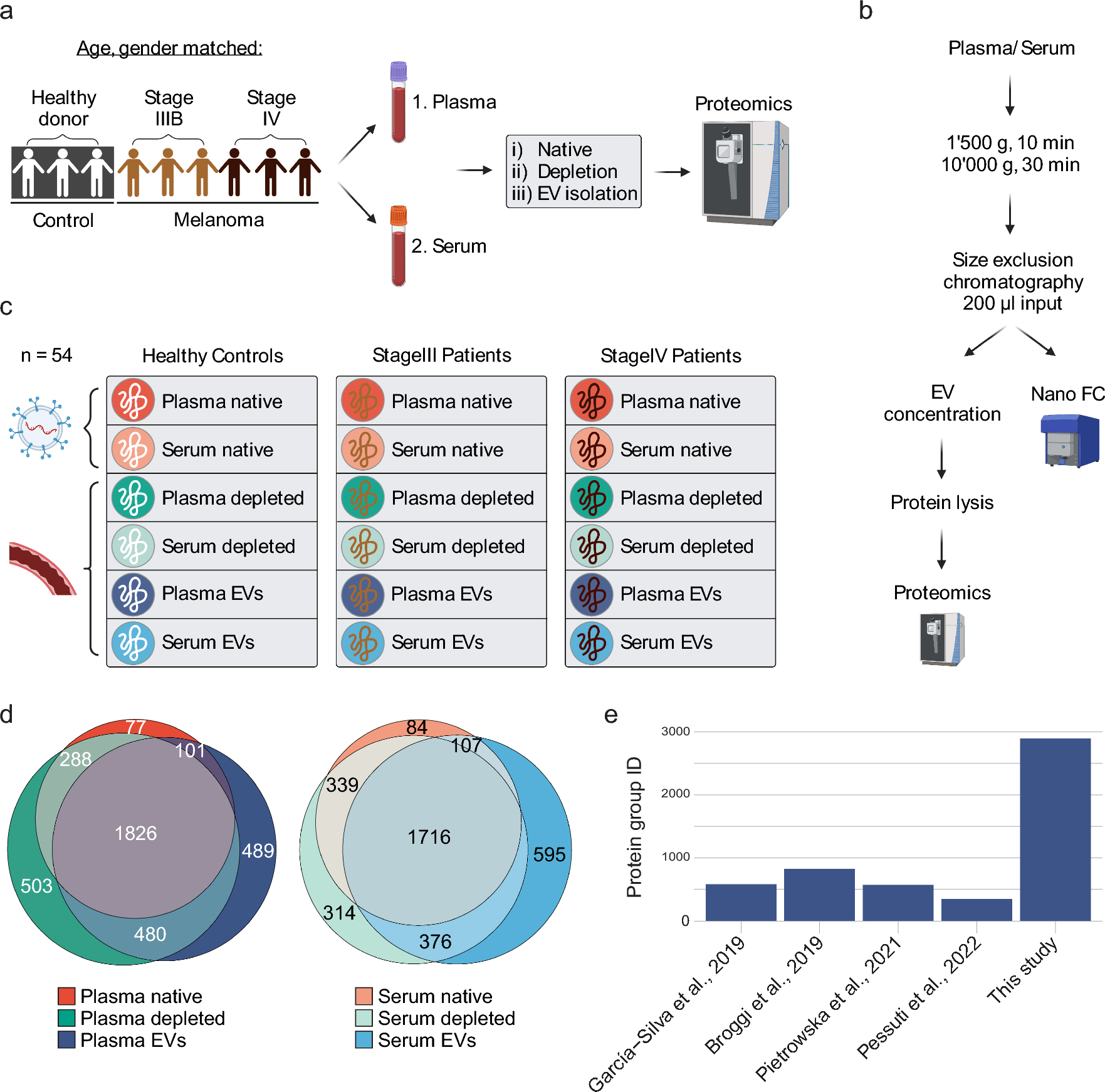
Complement activation and regulationThe complement system is an essential component of the innate immune system. It has been evolutionarily preserved for hundreds of millions of years and comprises roughly 30 membrane-bound and soluble proteins (Fig. 1) [10, 11]. The complement system is activated by three distinct pathways that occur both on pathogenic surfaces and in plasma (Fig. 1) [1, 12]. The classical pathway is primarily triggered by antigen-bound antibodies. Specifically, and the Fc regions of these activated antibodies (primarily IgM and IgG) bind to C1q, which initiates the complement classical pathway [13,14,15]. Human IgG3 and IgG1 bind and activate C1 readily, whereas IgG2 does so poorly, and IgG4 exhibits no activity [14,15,16]. The alternative pathway can be stimulated by attachment of C3b, a cleavage product of complement component 3 (C3), to foreign particles and damaged tissue, or by spontaneous cleavage of C3. The mannose-binding lectin (MBL) pathway is initiated when the plasma MBL protein complexes with MBL-associated serine protease-1 and -2 (MASP-1/2), which in turn binds to microbial surface oligosaccharides and acetylated residues (Fig. 1) [17].
Figure 1.
Adapted from: Lim et al. Blood Rev. (2023). Generated in Microsoft Powerpoint
Complement (C) activation, regulation, and C-targeted therapeutics: Complement is activated by classical. Lectin and alternative pathways, which leads to forming the complement activation bi-products such as C3a, C5a, and membrane attack complex (MAC). Complement activation is restricted by an array of host complement regulators. The therapeutics listed include complement-targeted therapy used in clinical trials for the treatment of COVID-19. FI Factor I, FH, Factor H, FP Properdin, C1INH, C1 esterase inhibitor, LPS lipopolysaccharide, MLB Mannose-Binding Lectin, FCN Ficolin, MASP Mannose-Associated Serine Protease.
The three extracellular pathways (classical, alternative, and MBL) coalesce via the activation of C3 convertase and the cleavage of C3 to C3a and C3b [1, 12, 18]. This engenders a cascade of cleavage and activation events, including the cleavage of C5 to C5a and C5b via a C3b complex that culminates in the formation of the membrane attack complex (MAC; Fig. 1) [1, 12, 18]. MAC formation begins with the sequential recruitment of C6, C7, and C8 to C5b. The MAC embeds itself in the phospholipid bilayer, and C8 induces the polymerization of C9 molecules to form a pore-like structure. The MAC is a macromolecular pore capable of inserting itself into cell membranes and lysing foreign pathogens, and heterologous cells. Under certain pathological conditions such as PNH, the loss of complement regulators on erythrocytes leads to lytic MAC formation which can cause hemolysis [12, 19,20,21]. Additionally, the formation of MAC at sublytic concentrations in a cell membrane of nuclear cells such as monocytes and endothelial cells can stimulate signaling cascades [22,23,24,25,26,27,28,29,30] that lead to the activation of monocytes and mediate inflammation on blood vessel without the lysis of the cells [2, 31,32,33,34]. In addition to complement activation on cell surfaces and in serum, recent evidence points towards intracellular complement activation via local production, endocytosis, and phagocytosis [35, 36]. C3 and C5 cleavage has been observed in various immune cells, including lymphocytes, monocytes, and neutrophils, and may also have a signaling function [37].
To prevent excessive complement-mediated damage, several plasma and membrane-bound protein regulators have evolved to attenuate and restrict the complement system at different stages of activation (Fig. 1) [1, 12, 18]. Plasma factor I (fI) controls the production of active C3b by cleaving C3b into inactive iC3b and C3d [1, 12, 18] (Fig. 1). Factor H, also soluble, regulates the alternative pathway by accelerating the decay of C3 convertase and is a cofactor for factor-I-mediated inactivation of C3b [38]. The soluble C1 inhibitor regulates the classical pathway upstream [1, 12, 18] (Fig. 1). Membrane-bound protein regulators consist of complement receptor 1 (CR1), membrane cofactor protein (MCP) CD46, decay-accelerated factor (DAF) CD55, and CD59 (Fig. 1). These regulators are expressed on host cell membranes and protect the host from complement attack by inhibiting upstream complement convertases, deactivating complement products, and restricting the formation of MAC by inhibiting complement pathway activation at varying levels of the cascade [1, 12, 18]. For example, CD59 inhibits the formation of the MAC by directly binding to C8 and C9 and preventing the polymerization of C9, while CD55 accelerates the decay of C3 and C5 convertases [1, 11, 12, 18]. These regulators mediate a delicate balance between adaptive and toxic immune responses.
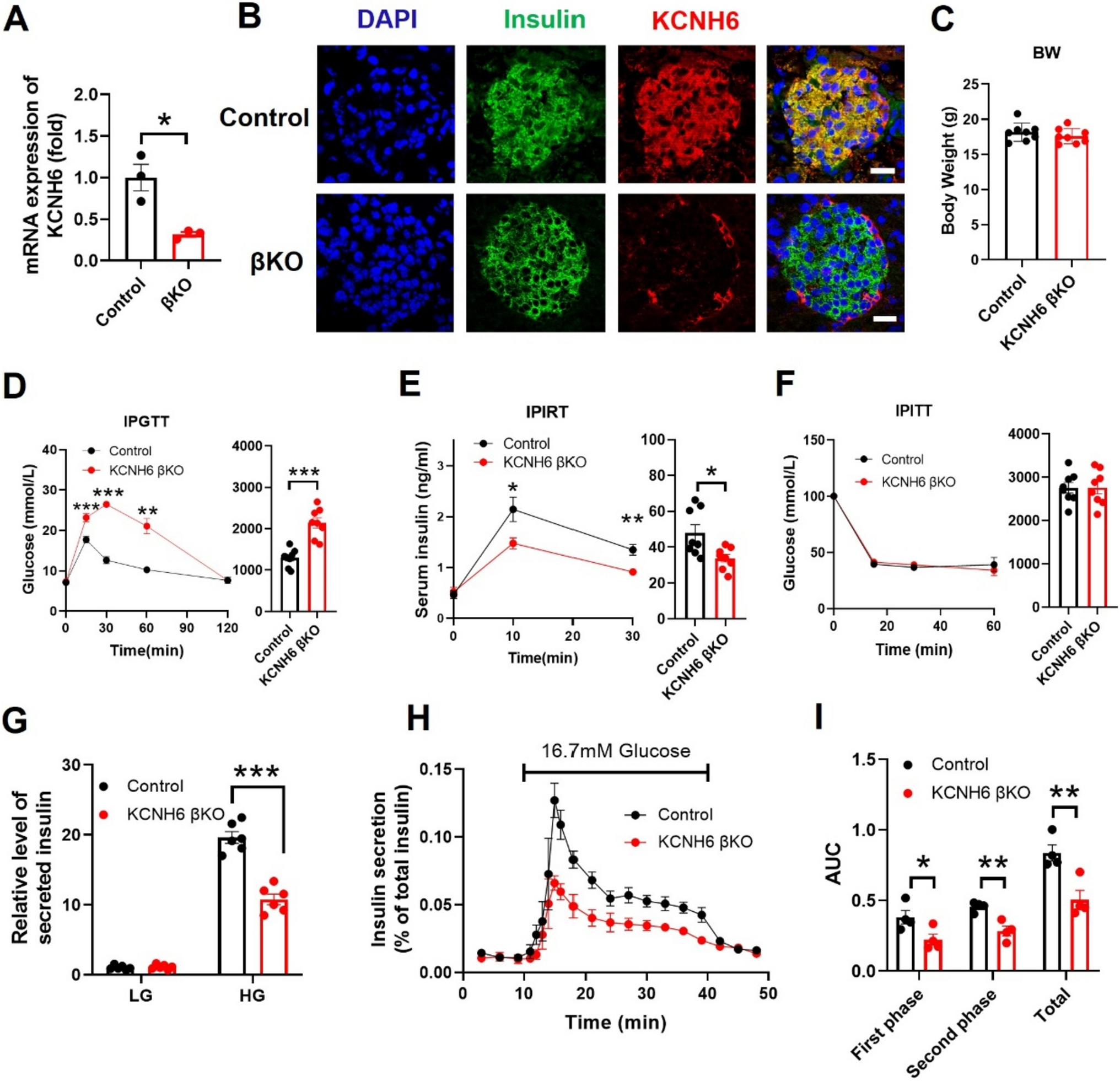
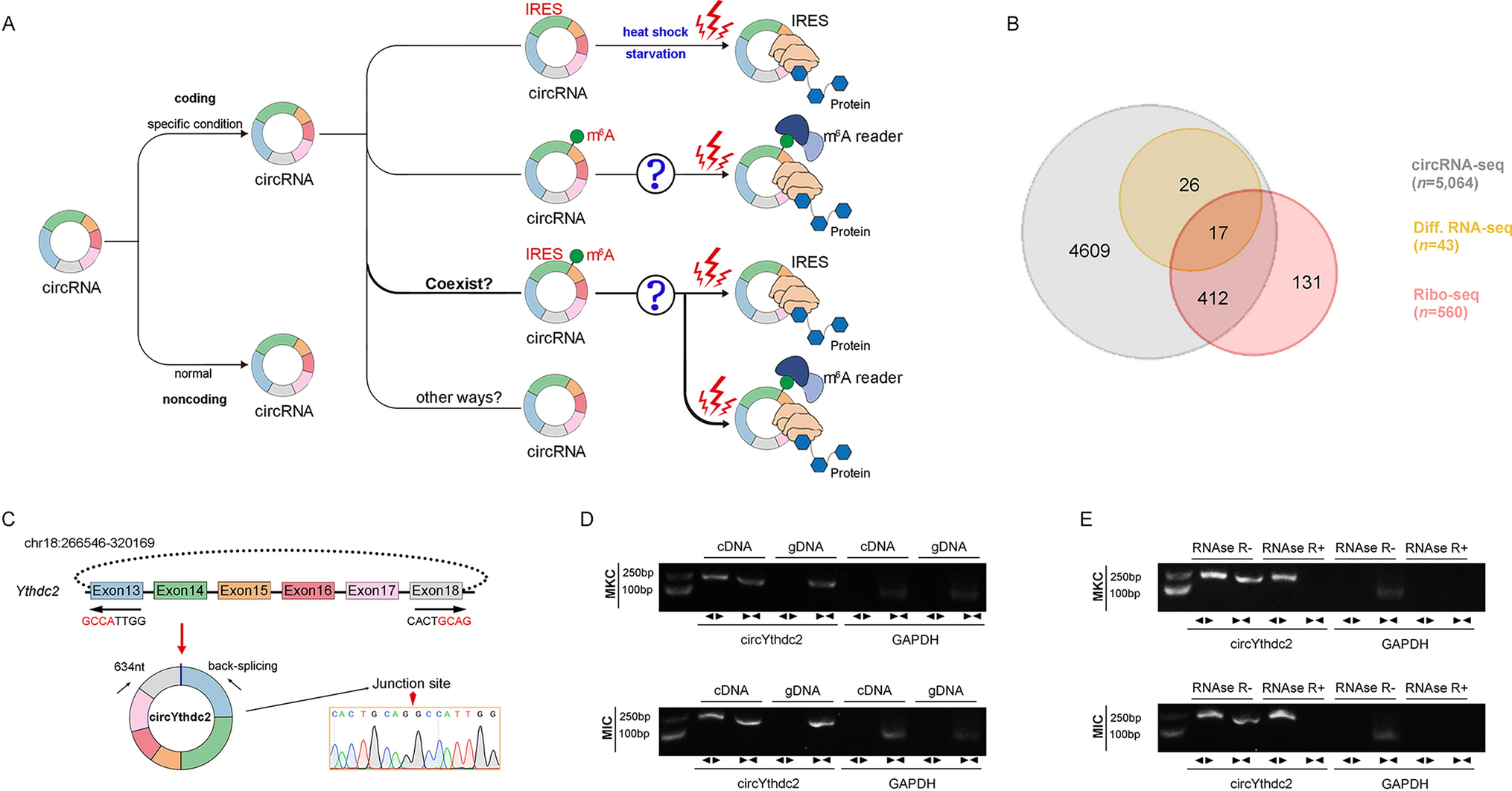
Proper regulation of the complement cascade is essential for a healthy immune response. Over- or underactivation is associated with disease pathologies; for example, although PNH and aHUS are distinct in terms of clinical manifestation, they both feature a mechanistic overactivation of the complement system via various malfunctioning complement regulators including CD55, CD59, Factor H, and Factor I [39, 40]. Figure 2 details examples of SARS-CoV-2-mediates complement activation and tissue damage, using various images available online under creative commons use licenses [41,42,43,44,45]. On the other hand, some tumors may develop resistance to complement-dependent cytotoxic (CDC) chemotherapy via the overexpression of CD59 (and consequently, the inhibition of MAC) [46, 47]. CD59 also plays a large role in virulence: Increased incorporation of CD59 into viral envelopes can protect against antibody-dependent complement-mediated lysis in HIV-1, cytomegalovirus, and herpes, among other pathogenic viruses [48,49,50,51,52,53]. Respiratory viruses such as SARS-CoV-2, influenza, and RSV all show evidence of heightened activation of the complement system upon infection, but the consequences of this activation vary dramatically (as reviewed below). The mechanisms underlying these distinct host responses to specific infections are unclear and warrant further investigation.
Fig. 2
SARS-CoV-2 mediates complement activation and tissue damage and causes the relapse of complement dysregulation-related diseses: SARS-CoV-2 cellular invasion is mediated by ACE2 receptor binding. Beyond viral mediated cell lysis, SARS-CoV-2 can cause damage to tissues in the lungs, endothelia, kidneys, and other organs via the direct activation (and the inhibition of regulation) of the complement system. SARS-CoV-2 can also mediate the relapse of complement dysregulation-mediated diseases such as atypical hemolytic uremic syndrome (aHUS), Complement-mediated thrombotic microangiopathy (TMA) and paroxysmal Nocturnal Hemoglobinuria (PNH) (highlighted in red). BALF bronchiolar Lavage Fluid, ARDS Acute Respiratory Distress Syndrome. S Protein Spike Protein, MAC Membrane Attack Complex, N Protein Nucleocapsid Protein. Images used were imported from online sources under creative commons use licenses or generated in Microsoft Powerpoint.
Increased complement activation is associated with severe COVID-19COVID-19-related morbidity and mortality are increasingly thought to be related to excessive immune response [54]. While the pathogenesis of severe COVID-19 varies, key manifestations include platelet activation, thrombosis, endothelial dysfunction, immune activation, and cytokine storm [55,56,57,58]. Proteomic analysis from tissue samples obtained early in the pandemic identified complement overactivation as one of the strongest indicators of COVID-19 infection. For instance, hospitalized COVID-19 patients have significantly higher levels of circulating sC5b-9 than similarly sick influenza patients and C5a and alternative pathway markers are associated with increased COVID-19 severity [59]. In addition to the alternative pathway, which is predominant earlier in the course of diseases, clinical evidence points to complement activation across the MBL and classical pathways as well [60].
While evidence increasingly points to COVID-19 being a systemic disease [61, 62], pathogenesis of acute COVID-19 infection begins in and most severely impacts the lungs via respiratory failure and inflammation. Early clinical observations indicate heightened complement deposition in various tissue, including C3 and C5b-9 on lung endothelium and C5a in bronchoalveolar lavage fluid (BALF) [63, 64]. Heightened complement activity may also play a critical role in thromboinflammation in severe COVID-19 patients via the platelet/neutrophil extracellular traps (NETs)/thrombin axis [56] (Fig. 2). Coagulation and inflammation drive ARDS and contribute to the severity of the disease, and ARDS has long been associated with complement activation [65,66,67]. Some evidence suggests that complement activity may contribute to COVID-19 severity in a dose-dependent manner, with higher concentrations of sC5b-9 (solubilized MAC), C5a, and C3 byproducts in particular being associated with respiratory failure [57, 61, 63, 68,69,70] (Fig. 2).
The kidneys follow as the second most commonly affected organ system in COVID-19 patients, with an estimated 25% of patients hospitalized due to COVID-19 developing acute kidney injury [71, 72] (Fig. 2). Again, biopsies reveal that these patients had similar/elevated levels of complement activation products in renal and glomerular arteries relative to those with other complement-related kidney pathologies [73]. In fact, immunofluorescence staining of samples from nine patients with fatal COVID-19-related acute respiratory failure revealed that C5b-9 deposition patterns in the kidneys were remarkably similar to those in the lungs [64](Fig. 2). The similarity in COVID-19-related complement pathologies across organs suggests systemic complement overactivation. The complement system has been implicated in other COVID-19-mediated organ damage as well. For instance, deposits of various complement proteins have been observed in the livers, hearts, and systemic and cutaneous vasculature of infected patients [62, 64, 74,
留言 (0)