
記住我

The monoculture model with Human cerebral microvascular endothelial cells (HBEC-5i) (from ATCC-Manassas, VA, USA) was conducted as described earlier [22]. HBEC-5i cells can grow monolayer on the polycarbonate membrane between the upper and lower compartments of Transwell plate, becoming an ideal BBB model in vitro with high TEER value and low permeability. The cells were cultured on 0.1% gelatin solution-coated (ATCC) flasks in Dulbecco's Modified Eagle Medium (DMEM)/F-12 medium (ATCC). They were supplemented with 10% fetal bovine serum (FBS, Gibco/Thermo Fisher, Waltham, MA, USA), 40 μg/mL endothelial cell growth supplement (ECGS, ATCC), 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Beyotime, Shanghai, China).
Lentivirus-Mediated Cav-1 RNA InterferenceWe established a stable Cav-1-silenced HBEC-5i cell line by infecting the cells with a lentivirus encoding Cav-1 shRNA (GenePharma, Shanghai, China) as before [21]. The sequence of targeted-Cav-1-RNAi used was ccACCTTCACTGTGACGAAAT. The negative vector contained a nonsense shRNA (5’- TTCTCCGAACGTGTCACGT -3’) to control for any non-RNAi-mediated effects. After synthesizing polymerase chain reaction (PCR) products based on these designs, they were cloned into the central part of a GV493 lentiviral vector. T293 cells were transfected with the packaging plasmid using GV493 for 48 h. The lentiviruses were then harvested with a collection of the supernatants, followed by concentrations and viral titers measurement. HBEC-5i cells were exposed to lentivirus-containing supernatant for 12 h. Stable transfectants were selected with puromycin (1 μg/ml) for 6 days and verified by real-time PCR.
Confocal Microscopic Analyses of GluN1 and Cav-1HBEC-5i cells grown on glass bottom cell culture dishes were fixed in 4% paraformaldehyde for 30 min, rinsed, permeabilized with 0.3% Triton X-100, rinsed a second time, and incubated with 3% bovine serum albumin in phosphate-buffered saline for 1 h. For double staining of GluN1 (1: 100, mouse monoclonal, #32–0500 in Thermo Fisher, Waltham, USA) and Cav-1(1: 400, rabbit monoclonal, #3267 T in CST, Boston, USA), cells were immunolabeled with a mixture of monoclonal anti-GluN1 and anti-Cav-1antibodies, washed, and treated with a mixture of Anti-rabbit IgG (H + L) F(ab')2 Fragment (Alexa Fluor®594 Conjugate, 1:500, #8889S in CST, Boston, USA) and Anti-mouse IgG (H + L) F(ab')2 Fragment (Alexa Fluor® 488 Conjugate, 1:1000, #4370 T in CST, Boston, USA). Images were obtained using Leica TCS SP8 Confocal Laser microscope.
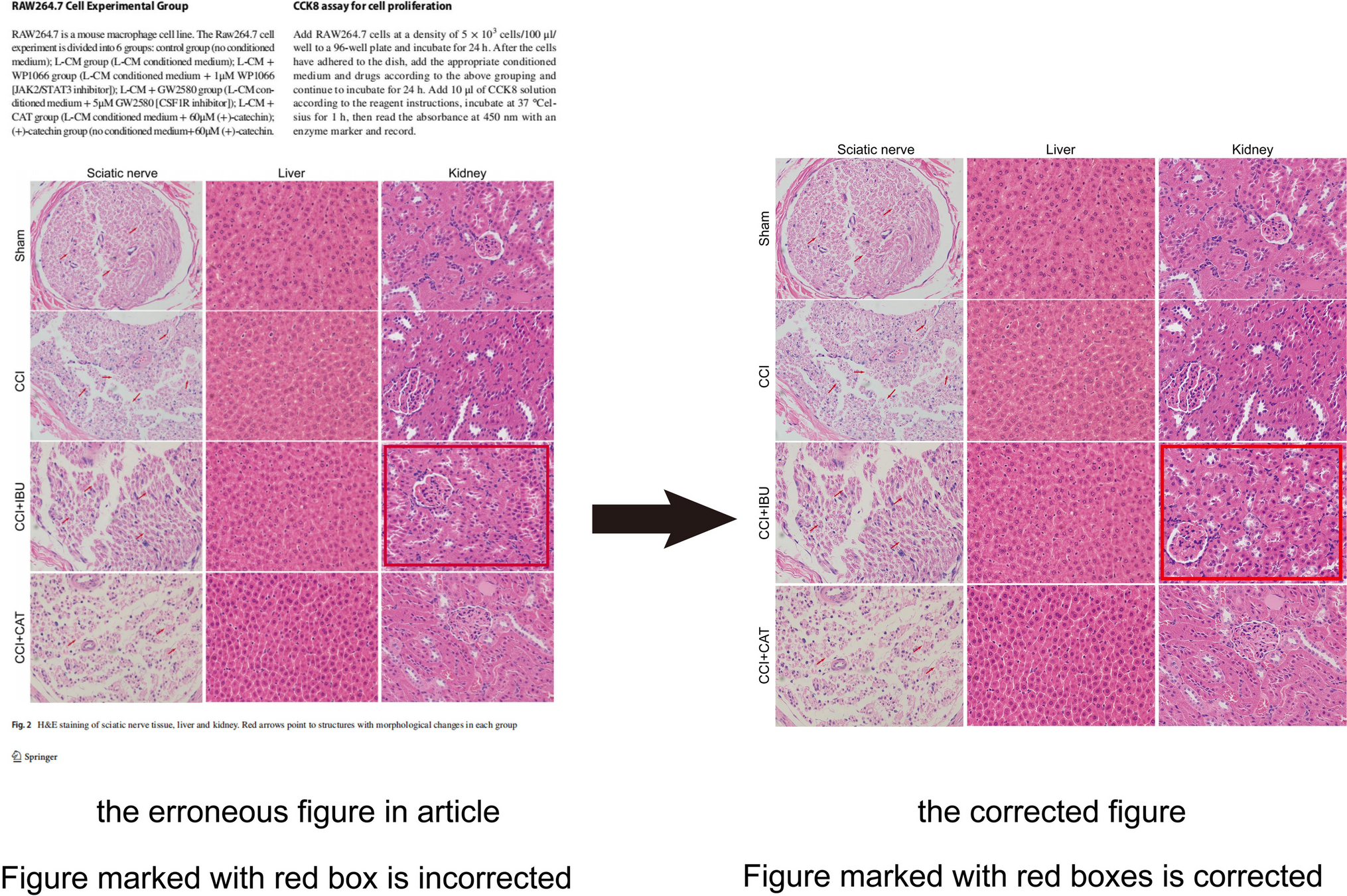
Experimental Design and Accessments of BBB Integrity in vitroThe interventions were carried out according to the following groups: 1) Blank control group; 2) NMDA (2.5 mM, HY-17551, MedChem Express, USA) group: HBEC-5i were incubated with NMDA alone for 24 h; 3) MK801 (10 μM, HY-15084B, MedChem Express, USA)/Daidzein (10 μM, HY-N0019, MedChem Express, USA)/LY294002 (10 μM, HY-10108, MedChem Express, USA) group: HBEC-5i were incubated with MK801/Daidzein/LY294002 for 24 h; 4) MK801/Daidzein/LY294002 + NMDA group: HBEC-5i cells were pretreated with MK801/Daidzein/LY294002 for 2 h, then incubated with NMDA for 24 h. The selected concentration of the drugs and their exposure time were confirmed in our previous study and other research literatures [21, 23,24,25]. The effects of MK801, Daidzein and LY294002 on the viability of HBEC-5i were assessed by a Cell Counting Kit-8 assay (Dojindo, Kumamoto, Japan) (shown in supplementary data). To further study the role of Cav-1 in the change of the Akt/mTOR pathway after NMDAR activation, the transfected cells were randomly grouped as followed: 1) LV-Ctrl shRNA: HBEC-5i were transduced with Ctrl shRNA lentiviral vectors; 2) NMDA + LV-Ctrl shRNA: HBEC-5i were transduced with Ctrl shRNA lentiviral vectors and incubated with NMDA for 24 h; 3) LV-Cav-1 sh RNA: HBEC-5i were transduced with Cav-1 shRNA lentiviral vectors; 4) NMDA + LV-Cav-1 shRNA: HBEC-5i were transduced with Cav-1 shRNA lentiviral vectors and incubated with NMDA for 24 h. Schematic diagram of different site intervention of Cav-1 related Akt/mTOR signal pathway is shown in Fig. 1.
Fig. 1
Schematic diagram of different site intervention of Cav-1 related Akt/mTOR signal pathway
The integrity of the BBB model was confirmed by measuring transendothelial electrical resistance (TEER) and using sodium fluorescein (SF) permeability assay. TEER values of monolayers of HBEC-5i were seeded on transwells then cultured for 2–3 days (the TEER value reached a plateau phase, TEER > 90 Ω*cm2) using an TEER measurement system -EVOM (ERS-2, Millipore, Burlington, MA, USA) [26]. Each TEER value was calculated by subtracting the resistance (Ω) value of a blank transwell and multiplying by the transwell surface area. SF permeability assay was measured as previously described [27]. Treated BBB models on cell culture inserts were tested for the flux of SF (10 µg/ml) in D-hanks solution. Meanwhile, the flux of cell-free inserts was measured to calculate the endothelial permeability coefficient (Pe).
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)Total RNA was extracted using the NucleoZol kit (MACHEREY–NAGEL, Germany). Then, 2 µg RNA was used for reverse transcription. The process of quantitative real-time PCR was performed in the Step One Plus Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). Relative gene expression was determined by the fluorescence intensity ratio of the target gene to GAPDH. Every group was repeated three times with a similar result. Primers were as follows:
Occludin F:5’-TCAGGGAATATCCACCTATCACTTCAG-3’.
R:5’-CATCAGCAGCAGCCATGTACTCTTCAC)
MMP9 F:5’-CCCTGGTCCTGGTGCTCCTG-3’
R:5’-CTGCCTGTCGGTGAGATTGGTTC-3’
Cav-1 F:5’-GCAGAACCAGAAGGGACACACAG-3’.
R:5’-CCAAAGAGGGCAGACAGCAAGC-3’
GAPDH F:5’-CAGGAGGCATTGCTGATGAT-3’
R:5’-GAAGGCTGGGGCTATTT-3'
Western Blot AnalysisTreated cells were prepared in Radio Immunoprecipitation Assay (RIPA) lysis buffer (Beyotime) for 30 min. After centrifugation at 12,000 rpm for 15 min, the supernatants were collected and protein concentrations were determined using BCA Protein Assay Kit (Beyotime). Samples were separated on SDS-PAGE gel, blotted onto PVDF membranes, then incubated with the respective primary antibodies at 4 °C overnight: GluN1 (1: 1000, mouse monoclonal, #32–0500 in Thermo Fisher, Waltham, USA), Occludin (1: 800, rabbit polyclonal, #27,260–1-AP in Proteintech Group, Chicago, IL, USA), MMP9 (1:800, #10,375–2-AP in Proteintech Group, Chicago, IL, USA), Cav-1 (1: 1000, rabbit monoclonal, #3267 T in CST, Boston, USA), p-Cav-1 (1: 1000, rabbit polyclonal, #3251 T in CST, Boston, USA), Akt (1: 1000, rabbit monoclonal, #4691 in CST, Boston, USA), p-Akt (1: 2000, rabbit monoclonal, #4060S in CST, Boston, USA), mTOR (1: 1000, rabbit monoclonal, #2983 in CST, Boston, USA), p-mTOR (1: 1000, rabbit monoclonal, #5536 in CST, Boston, USA), and GAPDH (1: 5000; rabbit polyclonal, 10,494–1-AP in Proteintech Group, Chicago, IL, USA). Then, samples were incubated with goat anti-rabbit fluorescent secondary antibody (1: 10,000; PA5-17,447 in Thermo Fisher Scientific) for 1 h at room temperature. The detected proteins were scanned and captured with the Odyssey Infrared Imaging System (LI-COR Biosciences). Bands were analyzed by ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Statistical AnalysisAll data were analyzed using SPSS version 22.0 (SPSS, Chicago, IL, USA) or GraphPad Prism software (version 9.3). The results were displayed as mean ± standard deviation (SD) and results were obtained from at least three separately performed experiments. One way ANOVA test was performed to determine whether there were significant differences between groups. P < 0.05 was considered to indicate statistical significance.
留言 (0)