
Remember me
The current strategy for clinical use of CDK4i/6i in patients with advanced ALM rests upon the genetic status of CDK pathway nodes, in part due to recent evidence that Cdk4 and/or P16INK4a copy number status may be of prognostic significance for ALM patients [7]; stemming from this study, only patients with CDK4 gain, CCND1 gain and/or CDKN2A loss were eligible for treatment with palbociclib [8]. In an independent analysis of a separate ALM patient cohort (n = 75 primary samples with survival information) [9], we find no significant correlation between the overall survival of ALM patients with wildtype CDK4 (n = 57) versus CDK4 gain (n = 16), or wildtype CDKN2A (n = 47) versus CDKN2A loss (n = 27) (Supplementary Fig. 1A, B).
We next characterized the relationship between gene copy number variations (CNVs) and baseline protein expression of CDK4 pathway nodes across a genetically diverse panel of human ALM and non-ALM cell lines (Fig. 1A, B, Supplementary Fig. 1C). In agreement with clinical observations, CDK4 pathway nodes (CDK4, CDK6, CCND1, CDKN2A, CDKN2B) were highly dysregulated across our ALM cell lines. CCND1, a key activator of CDK4 and CDK6, was elevated at the protein level in ALM versus non-ALM models (Fig. 1B). Notably, there was no consistent agreement between the copy number status and protein expression of CDK4, CDK6, or cyclin D1 in our ALM panel. ALM cell lines with CDK4, CDK6, or CCND1 copy number amplifications did not robustly display elevated CDK4, CDK6, or cyclin D1 protein expression relative to ALM models with normal gene copy numbers, respectively (Supplementary Fig. 1D–F). Analysis of the TCGA to understand the relationship between CNV status, mRNA level, and protein expression for cyclin D1 in patients (n = 89 with RPPA information available) with superficial spreading melanoma (CDK4 and CDK6 protein expression unavailable) also revealed no correlation between gene copy number or mRNA expression with protein expression (Supplementary Fig. 1G). This may serve as a cautionary note for the identification of ALM patients who may benefit from CDK4i/6i based solely on tumor sequencing, which may prevent patients with elevated CDK4/CDK6 protein expression but no clear evidence of copy number variation in CDK4/6 pathway genes from treatment.
Fig. 1: Genetic status of CDK4-pathway nodes does not predict protein expression or durability of CDK4/6 inhibition.
A Copy number variation and mutational status were assessed across a panel of ALM and non-ALM models. B Western blot showing basal expression of cell cycle proteins across a panel of ALM and non-ALM models. Shown in the right panel is a densitometric quantification of cyclin D1 expression. C Cells were treated with increasing concentrations of palbociclib for 24 h before Western blotting. D A panel of ALM models were treated with increasing concentrations of palbociclib, E ribociclib, or (F) abemaciclib for 72 h before cell numbers were quantified using MTT. Bars show S.E. mean. G A panel of ALM cell lines were treated with palbociclib for 3–4 weeks before colonies were fixed and stained with crystal violet. Photographs are representative of three independent experiments and relative clonogenic survival quantitation is shown to the right. H WM4324 cells were treated with palbociclib (500 nM) for the time shown before EdU incorporation and imaging to assess cell proliferation. Two sample t-test was used to compare means of any two groups’ Cyclin D expression, confluency and EdU incorporation in figure B, G, and H. *p < 0.05 and n = 3 unless otherwise stated throughout panels.
In agreement with the literature, treatment with the CDK4i/6i palbociclib, ribociclib, or abemaciclib potently inhibits CDK4/6 substrates (p-Rb, FOXM1), and E2F target proteins (PLK1, cyclin A) in a dose-dependent manner (Fig. 1C, Supplementary Fig. 1H, I). Despite potent CDK4/6 and E2F suppression, CDK4i/6i treatment elicited a predominately cytostatic effect, with a subpopulation of viable ALM cells remaining after short-term treatment over the course of three days in the presence of non-physiologically high concentrations of three clinically utilized CDK4i/6i’s palbociclib (>230 nM [10]) (Fig. 1D), ribociclib (>1 mM [11]) (Fig. 1E) and abemaciclib (>290 nM [12]) (Fig. 1F). When the treatment period is extended, therapy-resistant colonies continue to survive following >3-weeks of chronic exposure to CDK4i/6i (Fig. 1G). Accordingly, CDK4i/6i elicited an initial inhibition of proliferative capacity followed by reignition of cell cycle progression following long-term treatment as seen by EdU incorporation (Fig. 1H).
CDK6 protein expression has been recently demonstrated to indirectly predict for sensitivity to CDK4/6 inhibition in ER+ breast cancers, non-small cell lung carcinomas, colorectal carcinomas, and superficial spreading melanomas [13]. In contrast, an analysis of correlations for ALM sensitivity to CDK4i/6i suggest that CDK6 protein expression trends (p = 0.087) directly with palbociclib sensitivity in ALM (Supplementary Fig. 1J). The baseline protein expression of CDK4 (p = 0.72) and Cyclin D1 (p = 0.6) in therapy naïve cells did not correlate with ALM sensitivity to CDK4i/6i (Supplementary Fig. 1K, L). Alongside the first phase II clinical trial results of CDK4i/6i treatment in patients with advanced ALM, it was proposed following an analysis of 4 ALM patients that experienced clinical benefit and 5 ALM patients that did not experience clinical benefit to CDK4i/6i that low MCM7 expression and SH2B3 amplification could serve as predictive biomarkers of poor response to CDK4i/6i [8]. We did not observe significant relationships between MCM7 expression nor SH2B3 expression with sensitivity to any of the three clinically available CDK4i/6i tested (Supplementary Fig. 2A–C). We next put into context our CDK4i/6i sensitivity data with recent reports of potential oncogenic drivers of ALM. LZTR1, an adaptor for Cullin 3 ubiquitin ligase complexes, was proposed to serve as a driver of ALM aggressiveness [9]. No significant relationship emerged between LZTR1 expression and CDK4i/6i sensitivity in our data (Supplementary Fig. 2D). Further, CRKL, a signaling adaptor protein in pathways including the IGF1R-PI3K axis, was proposed to serve as an oncogenic driver of ALM [14]. We also did not observe a significant correlation between CRKL expression and CDK4i/6i sensitivity (Supplementary Fig. 2E). Altogether, these data suggest that baseline protein expression of CDK4, CDK6, and cyclin D1 do not correlate with the respective gene copy number status, and the sensitivity of therapy naive ALM cells to single-agent CDK4i/6i does not correlate to: (a) CDK4, CDK6, or CCND1 amplification or baseline protein expression, (b) reported CDK4i/6i sensitivity biomarkers (MCM7, SH2B3), or (c) proposed ALM oncogenic drivers (CRKL, LZTR1).
Loss of DUSP4 expression following CDK4i/6i promotes ERK activation and drives intrinsic resistance via cyclin D1Improvement in the efficacy of CDK4i/6i with inhibitors of the MAPK pathway has been reported in prostate adenocarcinoma [15], superficial spreading melanoma [16, 17], and uveal melanoma [18]. In the context of NRAS mutant superficial spreading melanoma, network modeling of tumor cells treated with MEKi has identified CDK4 as a key driver of therapy resistance [19]. However, the mechanistic connection between CDK4/6 activity and MAPK pathway signaling remains incompletely understood. We next investigated the MAPK pathway following CDK4i/6i in our ALM system and observe that although acute palbociclib treatment led to reduced activity of downstream CDK4/6 substrates (pRb, FOXM1) and E2F effectors (PLK1, cyclin A), a robust hyperactivation of MAPK signaling (pERK) and increased downstream cyclin D1 expression were observed across our ALM panel irrespective of NRAS or BRAF mutational or CNV status (Fig. 2A, Supplementary Fig. 1C). For example, CDK4i/6i-induced activation of ERK occurred in BRAFV600E mutant (WM4324), NRASQ61R mutant (WM4235), as well as BRAF/NRAS wildtype (YUSEEP) cells. Further, we observe equivalent hyperactivation of pERK and elevation of cyclin D following CDK4i/6i in AM cell lines with (a) wild type BRAF/NRAS copy numbers (WM4223), (b) NRAS copy number deletion (YUHIMO), (c) NRAS copy number gains (YUSEEP, WM4235), and (d) BRAF copy number gains (WM4324), which suggests that what we have observed cannot be solely explained by elevated NRAS/BRAF signaling given the heterogeneity of BRAF/NRAS mutational and CNV status across our panel. Hyperactivation of ERK and increased cyclin D1 expression were also observed following pharmacological inhibition of CDK4/6 with abemaciclib, ribociclib and genetic silencing of CDK4/6 (Supplementary Fig. 3A, B).
Fig. 2: ERK hyperactivation drives intrinsic CDK4i/6i resistance via cyclin D1.
A A panel of ALM cell lines were treated with palbociclib (500 nM, 24–72 h) before characterization by Western blotting (top panel). Densitometric analysis is shown in the bottom panel (n = 5 in each group). B Cells were treated with palbociclib (500 nM) and/or trametinib (10 nM for WM4324/WM4235, 1 nM for YUSEEP) for 72 h before characterization by Western blotting. C WM4324 spheroids were formed before implantation in collagen and treatment with palbociclib and/or trametinib for 72 h. Spheroids were subsequently stained with a viability stain and imaged by fluorescent microscopy (green indicates living cells, red indicates dead cells). D Cells were treated for up to 4 weeks with palbociclib (30 nM) and/or trametinib (0.3 nM) before colonies were fixed and stained with crystal violet. Quantification is shown in the right panel. E WM4235 cells were treated with palbociclib (500 nM) and/or trametinib (10 nM) for 24 h before subsequent staining with EdU. Shown in the panel to the right is quantification. F YUHIMO cells were stably transfected with either Empty Vector (EV) or DUSP4 (DUSP4 OE) in the presence or absence of palbociclib (500 nM) for 72 h before characterization by Western blotting. G YUHIMO cells were transfected with either EV or DUSP4 for 72 h before treatment with palbociclib (500 nM) for 72 h before proliferative capacity following EdU staining. H Cells were transfected with either non-specific siRNA (siNS) or siCCND1 in the presence or absence of palbociclib (500 nM) for 72 h before characterization by Western blotting. I WM4324 cells were transfected with either non-specific siNS or siCCND1 for 48 h before treatment with palbociclib (500 nM) for 6 h before proliferative capacity following EdU staining (n = 6 in each group). Paired t-test was used to calculate the significance of difference between treatment and control group in figure A. Ordinary One-Way ANOVA and Tukey test were used to perform multiple pairwise comparisons in figure C, E, and G. Two sample t-test was used to compare group means in figure D and I. *p < 0.05 and n = 3 unless otherwise stated throughout panels.
We next tested whether targeting the MAPK pathway with a MEK1/2 inhibitor (MEKi, trametinib) could ablate intrinsic CDK4i/6i resistance. Combination treatment with MEKi and CDK4i/6i decreases cell cycle proteins (FOXM1, p-Rb, cyclin D1, PLK1) to a greater extent than what was achievable by either compound as a single-agent (Fig. 2B), and increases apoptosis (cleaved PARP) relative to single-agent CDK4i/6i. Targeting the MAPK pathway at the level of ERK (VX-11e) also increased the capacity of CDK4i/6i to decrease cell cycle proteins (Supplementary Fig. 3C). Combination treatment with MEKi and CDK4i/6i increased the 3D cytotoxicity in ALM spheroids (Fig. 2C) relative to single-agent CDK4i/6i treatment alone. Further, concurrent MEKi + CDK4i/6i conferred the greatest antitumor durability in long-term colony formation assays (Fig. 2D), and most efficiently reduced the subpopulation of EdU+ ALM cells relative to single-agent treatment alone (Fig. 2E).
It was previously reported that reactivation of the MAPK pathway following BRAFi in BRAFV600E mutant superficial spreading melanomas was driven, in part, by reduced expression of proteins that negatively regulate the pathway, including members of the Sprouty (SPRY) dual specificity phosphatase (DUSP) family [20]. In our ALM cell line panel, we observe that CDK4i/6i treatment decreases protein expression of DUSP4 levels, not SPRY2 or DUSP6 (Fig. 2A, Supplementary Fig. 3D). We next tested the hypothesis that alterations in DUSP4 protein levels could be contributing to the hyperactivation of the MAPK pathway following CDK4i/6i. Overexpression of DUSP4 ablates the activation of ERK and induction of cyclin D1 expression following CDK4i/6i (Fig. 2F), overexpression of DUSP4 increases the antiproliferative efficacy of CDK4i/6i, as evidenced by the greatest reduction of cell cycle machinery and improved inhibition of EdU positivity relative to CDK4i/6i treatment alone (Fig. 2G), and overexpression of DUSP4 increases the ability of CDK4i/6i to suppress clonogenic outgrowth in long-term colony formation assays (Supplementary Fig. 3E). In contrast, genetic silencing of DUSP4 reduced the efficacy of CDK4i/6i (Supplementary Fig. 3F). Altogether, these results demonstrate the role CDK4i/6i-induced reduction of DUSP4 serves in ALM cell sensitivity to CDK4i/6i.
The MAPK pathway has been experimentally shown to regulate cyclin D1 in melanocytes and BRAFV600E superficial spreading melanoma [21], however, the connection between MAPK activity and cyclin D1 expression has not yet been established in ALM. Growing ALM cells in nutrient-replete media following serum starvation induces MAPK activity (pERK, pRSK) and downstream cyclin D1 expression, which could be blocked using MEKi or ERKi (VX-11e), demonstrating the MAPK pathway, at least in part, regulates cyclin D1 expression in ALM (Supplementary Fig. 3G). We next tested the hypothesis that ERK hyperactivation following CDK4i/6i preserves cellular proliferation by promoting cyclin D1 expression. Genetic silencing of cyclin D1 increased the cell cycle arrest potential of CDK4i/6i, evidenced by further decreased protein expression of pRb, PLK1, and FOXM1 (Fig. 2H). Genetic silencing of cyclin D1 also decreased the EdU+ subpopulation relative to what CDK4i/6i alone could achieve (Fig. 2I). In summary, these data suggest ALM cells adaptively escape single-agent CDK4i/6i by hyperactivating the MAPK pathway via, at least in part, reduced DUSP4 protein expression. The hyperactivation of ERK activity maintains the proliferative capacity of ALM cells treated with CDK4i/6i by promoting cyclin D1 expression.
ERK hyperactivation drives acquired resistance to CDK4i/6iWe next investigated the role of the MAPK pathway in acquired resistance to CDK4i/6i in ALMs. We generated ALM models with acquired CDK4i/6i-resistance (CDK-R) by treating therapy naïve ALM cell lines with increasing concentrations of palbociclib (10–500 nM) between 3 weeks and 2 months (Fig. 3A). CDK-R cells display reduced sensitivity to acute treatment with palbociclib (Fig. 3B), cross-resistance to ribociclib (Supplementary Fig. 4A), and regain their proliferative (EdU+ positivity) capacity (Fig. 3C). Notably, CDK-R cells display elevated phospho-ERK relative to parental cells (Fig. 3D), which functionally drives CDK4i/6i resistance as evidenced by induction of PARP-1 cleavage, reduction in cell cycle proteins, and decreased viability following combination treatment with MEKi (Fig. 3E, F). CDK-R also exhibited decreased sensitivity to CDK4i/6i in long-term colony formation assays relative to their respective parental cell lines (Fig. 3G, Supplementary Fig. 4B). Treatment with MEKi resensitized CDK-R cells to long-term treatment with CDK4i/6i (Fig. 3G), induced cytotoxicity in 3D CDK-R spheroids (Fig. 3H) and depleted the proliferative EdU+ subpopulation of CDK-R cells (Fig. 3I). Treatment of CDK-R cells with ERKi also induced significant suppression of cell cycle machinery (e.g., PLK1, FOXM1, cyclin D1) and induced apoptosis (e.g., PARP cleavage) (Supplementary Fig. 4C). Of note, we observed cell growth in select CDK-R cell lines was still affected by CDK4i/6i, which would suggest the existence of reversible mechanisms of acquired resistance to palbociclib that have been previously reported in cholangiocarcinoma cells [22].
Fig. 3: ALMs acquire resistance to CDK4i/6i via ERK hyperactivation.
A ALM cell lines were treated with increasing concentrations of palbociclib (50–500 nM; up to 2 months) to generate cells with acquired resistance (CDK-R). B Parental and CDK-R cells were treated with palbociclib (100 nM; 72 h) before cell number was quantified by MTT. C Proliferative capacity was assessed in WM4235 cells treated with palbociclib (500 nM; 72 h) and WM4235-CDK-R by EdU staining. Quantitation is shown in the right panel. D Parental and CDK-R pairs were characterized by Western blotting. E CDK-R cells were treated with trametinib (10 nM; 24 h) while in the constant presence of palbociclib (500 nM) before Western blotting characterization. F CDK-R cells were treated with trametinib (10 nM; 72 h) before cell number was quantified by MTT. G CDK-R cells were treated with palbociclib (500 nM) and/or trametinib (1 nM) for up to 4 weeks before colonies were fixed and stained with crystal violet. Quantification is shown in the lower panel. H YUSEEP-CDK-R spheroids were generated, implanted in collagen, and treated with palbociclib +/− trametinib for 72 h before viability staining and imaging (n = 4 for each condition). I WM4235-CDK-R cells were treated with palbociclib (500 nM) and/or trametinib (10 nM) for 24 h before staining with EdU. Quantitation is shown in the bottom left panel (n = 4 for each condition). J Cells were treated with siCCND1 in the presence of palbociclib (500 nM) for protein lysate was immunoblotted. K Cells were treated as in (J) before staining with EdU. Two sample t-test was used for pairwise comparisons throughout panels. *p < 0.05 and n = 3 unless otherwise stated.
Activation of ERK in the context of acquired CDK4i/6i-resistance has not been reported in melanoma, however evidence in the breast cancer literature proposes a role for de novo HER2 mutations in estrogen receptor positive (ER+) breast cancers [23]. We sequenced for HER2 mutations and copy number variations (CNVs) in our CDK-R models with acquired CDK4i/6i resistance versus their respective therapy naïve parental, and observe no new HER2 mutations or copy number gains (Supplementary Fig. 4D). Heterozygous RB1 loss was also recently reported as a biomarker for CDK4i/6i resistance in ER+ breast cancer. We observe RB1 deletion in one of our CDK-R models (YUHIMO-CDK-R), but no evidence of RB1 inactivating mutations across our CDK-R models relative to their respective parentals. In addition, evidence in hormone receptor positive (HR+) breast cancers suggest potential roles for upstream receptor overexpression (FGFR2) and de novo mutations in RAS, AKT1, AURKA, CCNE2, and/or ERBB2 in the activation of ERK following acquired CDK4i/6i resistance [24]. We sequenced for AKT1, FGFR2, FGFR3, FGFR4, and NRAS mutations and CNVs in our CDK-R models versus their respective therapy naïve parental counterparts, and observe no new mutations in these genes. We observed a copy number gain of NRAS in the WM4324 CDK-R cells versus its respective parental. No other new copy number gains were observed (Supplementary Fig. 4D).
In contrast to what we observed in therapy naïve cells treated with CDK4i/6i, three out of the four CDK-R cell lines expressed similar cyclin D1 levels relative to their respective parentals (Fig. 3D), which suggest rewiring of cyclin D1 expression may occur over time in the context of chronic treatment. However, genetic silencing of cyclin D1 also resensitized CDK-R cells to palbociclib as evidenced by reduced expression of pRb, FOXM1, and PLK1 (Fig. 3J) and depletion of EdU+ cells (Fig. 3K). Altogether, these results indicate that hyperactivation of the MAPK pathway drives acquired CDK4/6 inhibitor resistance and targeting MEK1/2 or cyclin D1 can reinforce CDK4i/6i efficacy.
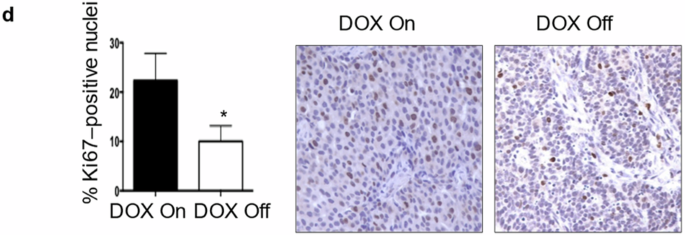
MEKi increases the in vivo efficacy of CDK4i/6i in therapy naïve and acquired CDK4i/6i-resistant ALM cellsTo assess the utility of targeting MEK to increase the in vivo efficacy of CDK4i/6i, we implanted the WM4223 patient-derived xenograft (PDX) model, derived from a biopsy of a metastatic ALM that originated in the left foot of a 73 year old male patient, into NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice (Fig. 4A). After 1–2 weeks, tumors were palpable and mice were treated via oral gavage with vehicle control, palbociclib (25 mg/kg), trametinib (0.3 mg/kg) or the combination of palbociclib plus trametinib, which was well tolerated (Supplementary Fig. 5A). Palbociclib and trametinib each conferred significant anti-tumor activity as single-agent treatments; however, the greatest therapeutic benefit was observed in mice receiving combination palbociclib plus trametinib treatment (Fig. 4B, Supplementary Fig. 5B). The ALM cell line YUSEEP, derived from the left heel, was also implanted in NSG mice and treated with vehicle control, palbociclib, trametinib, of the combination of palbociclib plus trametinib once tumors were palpable. Combination palbociclib plus trametinib treatment again conferred significantly greater antitumor activity relative to what could be accomplished by the single-agents alone (Fig. 4C, Supplementary Fig. 5C, D). Concurrent treatment with palbociclib and trametinib resulted in the greatest inhibition of cell cycle machinery (i.e., pRb, FOXM1, PLK1, cyclin D1) in lysate collected from a subset of tumor-bearing mice sacrificed after 3 days of treatment (Supplementary Fig. 5E) and conferred the greatest decrease of Ki67 staining in tumor tissue (Fig. 4D). At treatment endpoint for the WM4223 in vivo study (day 50), tumor tissue was characterized by reverse-phase protein array (RPPA) to identify the mechanism(s) of action underlying the long-term therapeutic efficacy of MEKi + CDK4i/6i relative to single-agent therapy (Fig. 4E). A total of 46 proteins were significantly differentially expressed between vehicle control tumors and combination palbociclib plus trametinib treated tumors that were not observed in the single-agent treated tumors (Supplementary Table). Interestingly, a signature indicative of reduced DNA repair capacity (decreased CENP-A, PARP and RPA32 protein expression) correlated with increased double strand DNA breaks in tumors treated with combination palbociclib plus trametinib (Fig. 4F, Supplementary Fig. 5F). Combination palbociclib plus trametinib treatment also resulted in the greatest cell cycle arrest signature (as seen by decreased cyclin B1, PLK1 and E2F1 protein expression (Fig. 4G)), and most significant induction of apoptosis (increased BAK, BID, BIM, caspase 7 cleavage, and reduced BCL2A1 protein expression) (Fig. 4H). In agreement, treatment of ALM cells with combination palbociclib plus trametinib for 10 days in vitro displayed increased DNA damage, BIM expression and cleavage of caspase 7 (Supplementary Fig. 5G).
Fig. 4: MEKi increases the in vivo efficacy of CDK4i/6i in therapy naïve and acquired CDK4i/6i-resistant ALM models.
A Schematic detailing the therapy naïve trial strategy. B Tumor growth curves of NSG mice implanted with the ALM PDX WM4223 and treated with vehicle control, palbociclib, trametinib, or the combination of palbociclib plus trametinib via oral gavage. C Tumor growth curves of NSG mice implanted with YUSEEP cells and treated with vehicle control, palbociclib, trametinib, or the combination of palbociclib plus trametinib via oral gavage. D IHC staining for Ki67 in WM4223 PDX tumor tissue from mice treated for 3 days with vehicle control, palbociclib, trametinib, or the combination of palbociclib + trametinib. E Heatmap of significant differentially expressed proteins between the palbociclib and control arm, the trametinib and control arm, and the combination palbociclib plus trametinib and control arm. F Plots depicting differentially expressed DNA repair and damage proteins from the data in (E), (G) Plots depicting differentially expressed cell cycle proteins from the data in (E), (H) Plots depicting differentially expressed apoptosis proteins from the data in (E). I Schematic detailing the acquired CDK4i/6i-resistant trial strategy. J Tumor growth curves of NSG mice implanted with the YUSEEP-CDK-R cells and treated with palbociclib, trametinib, or the combination of palbociclib plus trametinib via oral gavage. K Graphic summary of the manuscript findings. Wilcoxon signed-rank test was used to calculate significance when comparing tumor growth curves in figure B, C and G, 13, 12 and 24 time points are involved respectively, and n = 3 for each time point. Two sample t-test was used in figure F, G and H (n = 4 for control, n = 4 for Palbo, n = 3 for Trametinib, n = 4 for Palbo + Trametinib). *p < 0.05 unless otherwise stated.
To assess the utility of targeting MEK to overcome acquired resistance to CDK4i/6i in vivo, YUSEEP-CDK-R cells chronically treated with palbociclib in vitro were implanted in NSG mice (Fig. 4I). In line with observations of reversible (non-heritable) mechanisms of acquired resistance to palbociclib [22], YUSEEP-CDK-R cells that expanded in vivo during a >3 week drug holiday again exhibited sensitivity to single-agent palbociclib (Fig. 4J). Although vehicle treated YUSEEP-CDK-R tumors exhibited a greater growth rate relative to palbociclib treated, the vehicle treated YUSEEP-CDK-R tumors grew slower than the vehicle treated parental YUSEEP tumors (Fig. 4C). Treatment with MEKi significantly blunted tumor growth of CDK4i/6i-resistant ALM cells, and the combination of CDK4i/6i + MEKi resulted in the greatest antitumor activity and decrease in cell cycle proteins (pRb, cyclin D, PLK1, FOXM1) relative to what could be achieved by the single-agents alone (Fig. 4J, Supplementary Figure H, I, J). These findings indicate that continuous pressure from CDK4i/6i is required to maintain maximal vulnerability to MEKi. Altogether, these results underscore the importance of the MAPK pathway in driving intrinsic and acquired CDK4i/6i resistance in ALM and the translational potential of MEKi to increase the in vivo antitumor activity of CDK4i/6i against therapy naïve and CDK4i/6i-resistant ALMs, via increased DNA damage, cell arrest and tumor cell death (Fig. 4K).
Comments (0)