
Remember me






Transthyretin cardiac amyloidosis (ATTR-CA), also known as transthyretin amyloid cardiomyopathy,1 is one of the 2 most common types of cardiac amyloidosis (CA), and advanced cardiac amyloidosis is physiologically characterized as a restrictive cardiomyopathy.2 Amyloidosis is a systemic disease characterized by the misfolding of protein structures to form insoluble abnormal amyloid fibrils that are deposited in tissues and cause organ dysfunction.3 There are more than 30 types of proteins that form amyloid fibrils and cause amyloidosis. The most common type of protein that causes cardiac amyloidosis is immunoglobulin light-chain cardiac amyloidosis (AL-CA), which is caused by the clonal plasma cells secreting immunoglobulin light chain (AL), and transthyretin (TTR), a protein produced by the liver (formerly known as prealbumin) that causes ATTR-CA.4–6
TTR amyloidosis (ATTR) is divided into hereditary/variants (ATTRv) and wild type (ATTRwt) based on whether TTR has a gene mutation.7 More than 150 mutations in the TTR genes have been shown to cause ATTRv,8 and Val30Met is the most common mutation, associated primarily with neuropathy. It is endemic in Portugal, Japan, Sweden, and Majorca.9 The 4 mutations, namely Val122Ile, Leu111Met, Thr60Ala, and Ile68Leu, represent the predominant causative variants of ATTR-CA. Val122Ile is primarily prevalent among individuals of African American descent while Thr60Ala is more frequently observed in individuals of Irish heritage. The remaining 2 mutations are more commonly encountered in populations from Denmark and Italy.10 The clinical phenotypes of ATTRv amyloidosis include primary cardiomyopathy, polyneuropathy, and mixed type.11 ATTRwt is the transthyretin gene sequence that is normal,12 late-onset, predominantly male syndrome of cardiac amyloidosis,13 which mainly impairs the heart, kidney, thyroid, tendon and ligament tissues (hand, root canal, and ligamentum flavum), lung, and peripheral nerves, common in men older than 60 years, aging may contribute to its onset.14 Despite underdiagnosis and missed diagnosis, more than 50,000 patients have ATTRv amyloidosis worldwide.15,16 The 2016 ATTR Large Multicenter Study of THAOS (Clinical Trial Number NCT00628745) showed a high proportion of patients with ATTRv worldwide, with patients with ATTRwt accounting for only 12%.17 Transthyretin amyloidosis also has extracardiac manifestations, but its survival mainly depends on cardiac involvement, which is inferred to be one of the causes of heart failure in the elderly.18 Patients with ATTR-CA have a poor quality of life, a high rate of misdiagnosis, and low survival rates after diagnosis.1 Traditional biomarkers of heart failure, such as natriuretic peptide and cardiac troponin, are associated with risk but not specific, and ATTR-CA–specific biomarkers are currently undergoing clinical trials.19 40% of patients with ATTR-CA experience more than 4 years of repeated hospitalizations before receiving a definitive diagnosis.18 The median survival after diagnosis is 2.6 years for ATTRv-CA and 3.6–4.8 years for ATTRwt-CA.18,20 In addition, 13% of patients with preserved ejection fraction (HFpEF) is caused by ATTRwt amyloid.21Table 1 presents the clinical features. In recent years, there has been an exponential increase in the incidence and prevalence of CA, especially ATTR amyloidosis. Currently, patients with ATTR-CA show a more favorable cardiovascular prognosis than patients with AL-CA, which is associated with therapeutic agents that have been developed in recent years.22
TABLE 1. - Clinical Characteristics of ATTRv Versus ATTRwt Age at Symptom onset (y) Percentage of Men (%) Underlying Condition Duration of Symptoms before Diagnosis (y) Median Survival Time after Diagnosis (y) Organs or Systems Involved ATTRv >20 70–80 TTR mutation 2.8–4.3 2.5–3.5 (when heart is involved); 8–10 (when only nerves are involved) Heart, peripheral nerves, kidneys, eyes (vitreous opacity) ATTRwt >60 85–90 Age ∼2 3.6–4.8 Heart, muscle (carpal tunnel syndrome, conical stenosis, and biceps rupture), occasional peripheral nerve ATTRv, Genetic/mutant transthyretin amyloidosis; ATTRwt, wild-type transthyretin amyloidosis.14,18,20,23The contemporary approach to the diagnosis and treatment of ATTR-CA involves a sequential process. Clinicians must begin by assessing the likelihood of ATTR-CA, relying on clinical features associated with the cardiac amyloidosis phenotype.24 Patients displaying significant indications should proceed to the diagnostic phase. The initial step involves testing for monoclonal proteins. If the serum-free light-chain κ λ ratio falls below 0.26 or exceeds 1.65 or if monoclonal proteins are detected in either blood or urine, a tissue biopsy is then performed. This biopsy serves to confirm the presence of AL-CA, differentiate it from other forms of cardiac amyloidosis, or rule out amyloidosis altogether.1 If no monoclonal protein is detected, a cardiac radionuclide (99Tcm-PYP) imaging test should be performed.25 The ATTR-CA is diagnosed when the myocardial uptake rate is greater than or equal to the rib uptake rate (grade 2 or above).26 Tissue biopsy is the gold standard for the diagnosis of amyloidosis. It is applied in monoclonal protein detection, confirming the presence of 99Tcm-PYP, cases of clinical suspicion of cardiac amyloidosis when 99Tcm-PYP imaging is negative or ambiguous, and where 99Tcm-PYP scanning is not available.27 The final step in the diagnostic process involves genetic testing. Patients diagnosed with ATTR-CA should undergo genetic testing to further classify their condition into either ATTRv CA (variant ATTR-CA) or ATTRwt CA (wild-type ATTR-CA).2 Currently, ATTR-CA treatment has the following 3 aspects: (1) Treatment of heart failure: For patients with ATTR-CA, diuretics are mainly used to relieve the volume load and reduce the symptoms of heart failure, the standard treatment drugs for patients with HFrEF recommended in the guidelines do not benefit patients with ATTR-CA.1,27 (2) The treatment of cardiac arrhythmia: AF is the most common arrhythmia in ATTR-CA. Anticoagulation therapy is required regardless of the CHA2DS2-VASc score.25,27 Patients with developing conduction abnormalities may be considered for permanent pacemaker implantation,1 with studies estimating the patient expected survival of >1 year after implantable cardiac defibrillators (ICD).27 (3) ATTR-CA–specific therapy (etiological therapy): TTR tetramer stabilizer therapy (tafamidis) is the only drug approved for ATTR-CA.25 Additional drugs are undergoing clinical development.
This article presents a review of the development and clinical applications of new drugs for the treatment of ATTR-CA. It explains the pathophysiology of transthyretin amyloidosis and the mechanism of action of these new therapies, as well as provides an overview of the clinical research progress of various emerging drugs.
PATHOGENESIS AND MECHANISM OF ACTION OF NEW DRUGSThe core of ATTR-CA pathogenesis is the dissociation of TTR tetramer, which is mainly produced by the liver. Normally, TTR is a soluble tetramer that does not form amyloid fibers. When a single nucleotide of the TTR gene is mutated (ATTRv) or due to aging-related changes (ATTRwt),14,17,28 the TTR tetramer is easy to dissociate into monomer and misfold, leading to the formation of amyloid fibers that deposit in organs and tissues, resulting in amyloidosis.29–31 This process is called transthyretin amyloidosis, also known as ATTR amyloidosis.5 ATTR amyloidosis has 2 phenotypes, peripheral neuropathy (ATTR-PN) and cardiomyopathy (ATTR-CA), which often overlap and may be related to the nature of TTR mutations or other factors.32 The situation becomes more challenging when the phenotypic expression of ATTR amyloidosis is primarily or only cardiac.9 Most studies have demonstrated that amyloid deposits are physically destructive and a major cause of organ dysfunction.5,33 However, there is also evidence that toxic prefibrillary amyloid is a significant factor in heart failure.5,13 Most patients with ATTR die of cardiogenic causes, including heart failure and sudden death.34 In the early stages, ATTR-CA is often clinically treated as heart failure with preserved ejection fraction (HFpEF), progressing to heart failure with reduced ejection fraction (HFrEF).6 However, classical heart failure medications do not delay disease progression or improve survival, except for diuretics that relieve symptoms.1 Only the treatment of the cause can effectively control disease progression and prolong survival. Liver transplantation was applied in the 1990s to treat ATTR to block the production of mutant TTR and prevent the progression of the disease, particularly the Val30Met mutation.35 In ATTR-CA, heart transplantation or combined heart–liver transplantation is a potential cure36 with a higher survival rate than liver transplant patients alone.37 After a successful liver transplantation, it is crucial to understand that organ damage and neuropathy typically remain irreversible. In certain instances, disease progression may still occur. Notably, the Val30Met subtype demonstrates an overall 5-year survival rate of approximately 100% while patients with non-Val30Met subtype exhibit a 5-year survival rate of 59%.38 Studies have reported that cardiac amyloidosis occurs after liver transplantation, and wild-type TTR is a major component of amyloid deposition because the wild-type TTR is deposited on the already variant amyloid fibrils that form new-deposition matrix.39 Moreover, wild-type TTR will continue to produce fibers in the patient, preferentially deposited in the heart, and the disease continues to develop, liver transplantation is less effective; thus, it does not resolve all types of ATTR-CA.1,37 In addition, patient tolerance to advanced disease, lack of donors, and subsequent lifelong immunosuppressive therapy impose limitations on clinical transplantation. Based on the pathophysiological mechanism of ATTR, current drug therapy strategies mainly include transthyretin stabilizer that stabilizes TTR tetramers, a transthyretin silencing agent that interferes with TTR synthesis at the gene level (also known as gene silencing agent or transthyretin synthesis inhibitor) that have been applied clinically.40,41 Examples include small interfering RNA (siRNA) and antisense oligonucleotide. Key therapies in development include gene editing technology (CRISPR/Cas9), which is expected to replace combined heart–liver transplantation,42 emerging drugs that degrade and destroy amyloid fibrils (monoclonal antibodies),43 and early development stages such as extractors/depressors, antiseeding therapies, and TTR aggregation inhibitors.41 A variety of drugs are effective at different stages of pathogenesis (Fig. 1), and these specific treatments bring more options and new life to patients.31,42,44
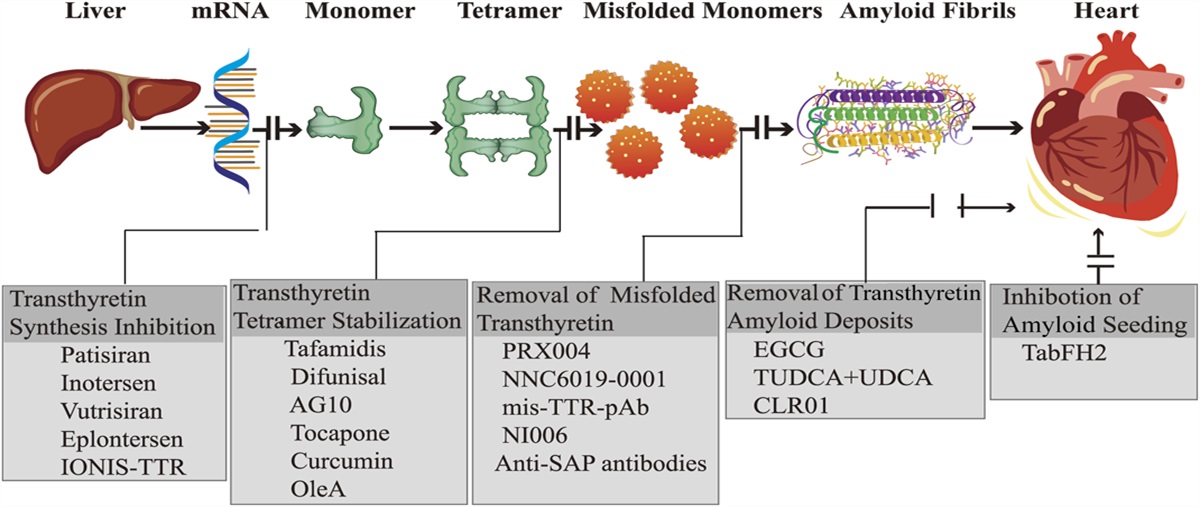
 FIGURE 1.:
FIGURE 1.: Transthyretin cardiac amyloidosis and mechanisms of drug action.
NEW DRUG DEVELOPMENT IN THE PRECLINICAL STAGEEmerging therapeutic approaches, including antiseeding therapy, are gaining significant research interest. In the realm of preclinical drug development, several promising candidates have surfaced. These include mis-TTR pAb (targeting amyloid fiber degradation/extraction), CLR 01 (an interrupter of transthyretin fiber aggregation), the novel TTR stabilizer curcumin, and OleA. In this context, we present a compilation of these preclinical-stage drugs and their essential data (Table 2), which presents a promising new direction in the treatment of cardiac amyloidosis.
TABLE 2. - Key Data From Preclinical Drug Studies Drug Name Type of Medication Researcher, Time Subject, Type of Experiment Primary Outcomes References mis-TTR pAb Amyloid fiber degradation/extraction Higaki et al (2016) Cell culture, molecular experiments Antibodies selectively bind to the monomers of TTR and target epitopes in the unnatural misfolded form, significantly inhibited TTR fibrils formation 45 CLR01 Methyl transferrin protein fiber aggregation interrupter Sinha et al (2011) Molecular experiments It binds to lysine residues, disrupt electrostatic and hydrophobic interaction to regulate nucleation, oligomerization and fibril elongation, and thereby inhibiting the aggregation and toxicity of various amyloid proteins; exposure to CLR 01 at concentrations above the concentration required for inhibition has no toxicity 46 Ferreira et al (2014) Cell culture and mouse models The TTR burden of the gastrointestinal tract and peripheral nervous system in ClR01-treated mice was significantly reduced, accompanied by aggregation-induced reduction in ER protein oxidation, stress response, and apoptosis 47 TabFH2 Amyloid seed inhibitor Saelices et al (2018, 2019) Cell culture, in vitro experiments TabFH2 completely inhibits TTR amyloid seeding, and TabFH2 inhibits TTR aggregation in a dose-dependent manner when the patient's amyloid fibril is seeded in vitro 49,51 Saelices et al (2018) A model of neuropathic ATTR in Drosophila melanogaster TabFH2 not only reduced TTR deposition in treated flies but also improved motor injuries in untreated flies 52 Curcumin TTR stabilizer Ferreira et al (2016) Mouse model Curcumin decreased transthyretin aggregate deposition and toxicity in the dorsal root ganglia and gastrointestinal, and also reshaped hemolytic amyloid substances in the tissue 55 OleA TTR stabilizer Bemporad et al (2022) Cell culture, molecular tests OleA interacted with TTR in different patterns to stabilize it and prevent it from dissociating into monomers and subsequently misfolding 57 Leri et al (2016) Cell culture OleA could break down preformed TTR mature fibril into the same nontoxic oligomeric-like intermediates 58Mis-TTR pAb is a conformational-specific antibody that selectively binds to the monomeric TTR or its misfolded conformation, inhibiting the formation of TTR fibrils in vitro. However, it does not recognize natural tetramers.45 Higak et al45 conducted a study describing the production of mis-TTR monoclonal antibodies that inhibit TTR fibrogenesis and induce antibody-dependent phagocytosis uptake of TTR aggregates in vitro.
Transthyretin Fiber Aggregation InterrupterCLR 01 is a lead compound of lysine-specific “molecular tweezers” critical for oligomerization and filament extension of nucleation by binding to lysine residues, disrupting electrostatic and hydrophobic interactions, thereby inhibiting the aggregation and toxicity of various amyloids, including TTR.46 Ferreira et al47 showed that CLR 01 regulates TTR self-assembly in the early stages and suppresses TTR-induced neurotoxicity. It is a process-specific and extensive inhibitor of amyloid formation.
Amyloid Protein Seeding Inhibitor (Antiseeding Therapy)TabFH2 inhibits seeding agents (peptide inhibitors), and a mixture of 2 optimized peptide inhibitors for F (TabF2) and H (TabH2), which cover the tips of F and H chains, which are aggregation-driven fragments of TTR,48 thereby inhibiting self-association and seeding. The name “Tab” refers to transthyroxine aggregation bloc, letters F and H refer to the target chain, and the number “2” depends on the originally designed peptide (number 2).49 The seeding of amyloid protein is the deposition of preformed amyloid fibrils in soft tissues or organs.50 Saelices et al49 found that amyloid fibril seeds derived from the patient's heart prompted the formation of amyloid fibrils by wild-type TTR in vitro. This seed was inhibited by peptides designed to complement the TTR fibril structure that can limit the growth of fibrils. In another study, Saelices et al51 found that the binding of TabFH2 to fibrin in vitro effectively inhibited the seeding of amyloid by blocking the self-binding of amyloid-driven chains F and H. At the same time, in another study by Saelices et al52 on the drosophila model carrying human ATTR, they found that TabFH2 not only reduced TTR deposition in flies but also improved the movement disorders observed in flies, the effect superior to diflunisal. TabFH2 antiseeding peptide inhibits amyloid aggregation and blocks the deposition of amyloid protein in body tissues. This affects fiber self-binding and subsequent polymerization, controlling the amyloid seeding process.53
TTR StabilizerCurcumin binds at the thyroxine site and interacts with TTR, stabilizing the TTR tetramer. In addition, it exerts regulatory control over TTR misfolding, promoting the formation of nontoxic protein aggregates, inhibiting aggregation, and directly catalyzing the disintegration of fibrillar structures.54 In addition, it clears TTR aggregates by endocytosis of fibroblasts and macrophages.54 Ferreira et al55 showed that when curcumin was used in familial amyloid polyneuropathy mice, it not only reduced TTR deposition and toxicity but also reshaped tissue affinity substances. Curcumin inhibited TTR aggregation in a dose-dependent manner and enhanced macrophage clearance of TTR aggregation in vitro. Curcumin is a promising drug candidate for protein misfolding neurological diseases,56 but it requires more clinical trials to prove its effectiveness in ATTR-CA.
OleA (Oleuropein aglycone) is a major polyphenol in extra virgin olive oil that can interact with TTR in different patterns, stabilizing the TTR tetramer structure and preventing the misfolding of its dissociated monomers.57 Leri et al58 conducted in-depth biophysical and morphological analysis of ATTR, with OleA reducing cytotoxicity that inhibits TTR amyloid aggregation by interfering with fiber aggregation, stability, and cytotoxicity to cardiac HL-1. Understanding this interaction at the molecular level can provide a basis for OleA treatment and prevention of various TTR amyloidosis.57
DRUG APPLICATION AND CLINICAL RESEARCH OF ATTR-CA IN THE MARKETCurrently, TTR stabilizers and TTR silencers have been proven to be effective against transthyretin cardiac amyloidosis, and some drugs have been approved for clinical application. In addition, several other drugs are in the early stages of development, such as gene-editing therapies, fiber degraders, and fiber aggregation–disrupting agents, most of which have shown good therapeutic outcomes in clinical trials. Here, we present pivotal clinical trials (see Table, Supplemental Digital Content 1, https://links.lww.com/JCVP/A990).
Transthyretin StabilizerTafamidis (Fx-1006A) is the first drug approved by the US Food and Drug Administration (FDA) for the treatment of ATTR-CA in 2019 and is the preferred clinically used drug. The 2022 AHA/ACC/HFSA Guidelines for Heart Failure Management classifies Tafamidis as a Class I recommended treatment for patients with ATTR-CA.2,59 Tafamidis binds to the thyroxine-binding site, inhibits the dissociation of TTR into monomers, stabilizes tetramers, and prevents the formation of amyloid fibers. Earlier studies: A Phase II open-label study demonstrated that tafamidis stabilized transthyretin variants, but the LV wall thickness was unchanged in most patients with ATTR-CA.60 A Phase II, multicenter study (NCT00694161)61 showed that tafamidis could slow disease progression, improve survival duration, and improve the rationale for tafamidis administration in ATTR-CA. The ATTR-ACT study (NCT01994889), which ended in 2018, was a phase III, international, multicenter, randomized, double-blind, controlled trial.62 It included 441 patients with ATTR-CA who were followed for 30 months. Initial data analysis showed that compared with placebo, tafamidis reduced cardiovascular-related hospitalization and all-cause mortality, slowed the rate of decline in quality of life and 6-min walk distance in the experimental group. This study also demonstrates that tafamidis treatment can reduce mortality and slow the rate of functional decline in various disease subtypes, including ATTRv-CA and ATTRwt-CA. Importantly, the observed effects are consistent across these different subtypes.63 The ATTR-ACT extension study predicted that tafamidis would more than double life expectancy and quality-adjusted life years in patients with ATTR-CA.64 It has also been concluded that both 20 and 80 mg of tafamidis effectively reduced the cardiovascular-related hospitalization and mortality rate in patients with ATTR-CA and 80 mg was the optimal choice.65 In addition, according to Rozenbaum et al,66 the ATTR-ACT study revealed significant benefits of tafamidis treatment. When compared with placebo, early tafamidis treatment led to a 52% reduction in the annual number of CVD-related hospitalizations. In addition, each hospitalization was shortened by an average of 1.14 days. In patients with grade I/II cardiac function, the annual number of CVD-related hospitalizations decreased by 3.96 days. Furthermore, there was a noteworthy 24% reduction in the frequency of CVD-related hospitalizations among NYHA class III patients who received tafamidis treatment.67 Late-stage clinical trials: Badr et al68 conducted a study on 54 patients with ATTR-CA to evaluate the effect of 9 months tafamidis treatment on functional ability through the cardiopulmonary exercise test (CPET). They found a significant improvement in physical activity ability at the last follow-up. Rettl et al69 demonstrated that a daily dose of 61 mg of tafamidis in patients with ATTR-CA effectively delayed the progression of extracellular volume dilatation and LV dilatation. It prevented further deterioration of LV function and influenced the cardiac structure, function, and clinical status. In another study by Hussain et al,70 an observational cohort analysis, it was concluded that tafamidis provided a clear survival benefit for patients within a community setting. This benefit extended to older individuals and those with more advanced disease, highlighting the broad effectiveness of tafamidis across different patient groups. Although tafamidis is a landmark breakthrough in the treatment of patients with ATTR-CA, further clinical research is needed to determine its benefit in late-stage ATTR-CA. Ongoing clinical trials include a Phase III study (NCT02791230) to assess the long-term safety of tafamidis in patients with ATTR-CA and a Phase Ⅳ study (NCT05489523) to explore the efficacy, safety, and pharmacokinetics of tafamidis in patients with transthyroxine-mediated amyloidosis after orthotopic cardiac transplantation.
Diflunisal, a nonsteroidal anti-inflammatory drug,71 stabilizes transthyretin tetramers, reduces the formation of amyloid fibers, and slows the progression of transthyretin amyloidosis. It has been shown to slow the polyneuropathic progression of hereditary ATTRv but has only been reported in a small group of patients with ATTR-CA.72 In the early days, Castan et al73 performed a survey study with a follow-up period of 0.9 ± 0.3 years, showing that diflunisal was well-tolerated at an oral dose of 250 mg diflunisal twice daily in patients with ATTR-CA included in this study. In a longitudinal, prospective, single-center study, it was found that sensitive markers of changes in cardiac function, such as LV wall thickness and NT-proBNP, remained stable in the experimental group, while both were increased in the control group, and this suggests that diflunisal may stop or slow the development of ATTR-CA disease progression.74 Later, Sekijima et al75 demonstrated the effectiveness and safety of diflunisal. The drug was well-tolerated, effectively stabilizing the TTR tetramer structure, and the clinical benefits persisted even after 2 years of treatment. Moreover, in the study by Ikram et al,72 diflunisal was well-tolerated in patients with ATTR-CA, and no patient died or was hospitalized due to heart failure during the average of 2 years of follow-up, and only 13% of patients experiencing gastrointestinal discomfort that could be prevented by food and proton pump inhibitors. In recent studies, Lohrmann et al76 reported that diflunisal can safely alleviate some parameters of cardiac function decline in patients with ATTR-CA 1 year after administration. It may serve as a viable and inexpensive alternative for patients with ATTR-CA who cannot afford or access FDA-approved therapies.
AG10 is a novel, effective, and selective oral transthyretin (TTR) stabilizer77 currently being developed for the treatment of ATTR. In the early days, Penchala et al78 reported that AG10 prevented the abnormal disintegration of V122I-TTR in the serum of patients with ATTRv-CA, showed high selectivity and potency of TTR in human plasma, lacked toxicity, and had good dynamic metabolism in rodents, all of which were confirmed in ATTR-CA treatment. A Phase I study by Fox et al79 showed that multiple and single doses of AG10 were well-tolerated with no potential safety risks and a high degree of TTR stability in healthy adult volunteers. Meanwhile, a phase II, randomized controlled study (NCT03458130)80 showed that AG10 was well-tolerated in patients with chronic heart failure with symptomatic ATTR-CA, TTR was almost completely stabilized at drug–target concentrations, and TTR levels returned to normal. AG10 is expected to be a safe and effective drug for the treatment of ATTR-CA. Safety and efficacy Phase III clinical trials (NCT03860935, NCT04988386) for AG10 are ongoing.
Tolcapone, an FDA-approved treatment for Parkinson's disease, binds tightly to the t4 binding site of amyloidosis-associated variants,81 restoring the tetramer stability and thereby inhibiting the aggregation of pathogenic proteins.82 It can also be used as an effective inhibitor of TTR aggregation, binding specifically to TTR and stabilizing native tetramers in humans and mice, and inhibiting the cytotoxicity of TTR. Sant'Anna et al83 showed that in vitro model experiments demonstrated that tolcapone is an effective inhibitor of TTR amyloid production, preventing early events and mutated TTR from inducing cytotoxicity.
Transthyretin SilencerThe transthyretin silencer mainly consists of 2 drugs: small interfering RNA and antisense oligonucleotide. Both of these drugs reduce intrahepatic TTR generation by preventing RNA messenger transcription.1
Patisiran is a small interfering RNA drug that is currently approved for use in patients with ATTR-PN. It is a lipid nanoparticle that is released after reaching hepatocytes and induces cleavage of TTR messenger RNA to block TTR protein production and mutation.84 Coelho et al85 conducted a Phase II clinical study that showed that patisiran can improve or stop polyneuropathy in patients with ATTR amyloidosis and improve their quality of life. Schmidt et al86 conducted a Phase III global study (NCT03862807) that showed the use of patisiran in patients with ATTRv amyloidosis after liver transplantation significantly reduced serum TTR and was well-tolerated. The APOLLO study is an international, multicenter, double-blind, randomized controlled Phase III clinical study (NCT01960348) for patients with ATTR-PN to evaluate the safety and efficacy of patisiran.87 Zhang et al88 reported on the pharmacokinetics (PK), pharmacodynamics (PD), and exposure-response analysis of the APOLLO trial. The maximum reduction in serum ATTR levels from baseline was 87.8% in patients treated with patisiran, and the pharmacokinetic exposure remained stable after continuous administration. Solomon et al84 conducted an analysis based on data from the APOLLO research, which shows that at 18 months, the patisiran regimen compared with the baseline and placebo groups improved including reductions in NT-proBNP, LV wall thickness, and in the overall cardiac longitudinal strain. The cardiac subgroup analysis of APOLLO showed that after 18 months of patisiran, the magnitude of LV longitudinal strain deterioration was significantly reduced compared with the placebo group, specifically the basal segment, demonstrating that patisiran had significantly stopped the progression of cardiomyopathy.89 The role of patisiran in delaying cardiac amyloidosis will be further validated in the ongoing APOLLO-B study (NCT03997383), which is a study population of patients with ATTR-CA.
Revusiran, a first-generation small interfering RNA that targets transthyretin and binds to n-acetylgalactosamine (GalNAc) ligand to facilitate delivery to liver cells through uptake of sialoglycoprotein receptors90 and is a first-generation GalNAc conjugate. A phase III study (NCT02319005) by Judge et al91 was terminated due to an observed mortality imbalance, leading to the discontinuation of drug development. Despite this setback, data from other large clinical studies showed the potential benefit of TTR-targeting siRNA in treating cardiac manifestations of the disease, thus advancing the study process of another TTR-targeting Galac-siRNA conjugate, vutrisiran (ALN-TTRsc02).
Vutrisiran (formerly ALN-TTRSC02) is a second-generation investigational RNAi therapy,92 a small interfering ribonucleotide drug, and a liver-directed treatment for transthyretin amyloidosis.93 The drug received approval in the United States in June 2022, and positive recommendations for stage 1 or stage 2 adult ATTRv-PN treatment were obtained in the EU in July of the same year. A phase I study evaluated the pharmacodynamics, pharmacokinetics, and safety of subcutaneous administration (5–300 mg) in healthy subjects, and treatment with vutrisiran achieved an effective and sustained reduction in TTR in a dose-dependent manner, with a mean maximum TTR reduction of 57%–97% lasting more than 90 days.93 The HELIOS-A, phase III, randomized study (NCT03759379) with 164 participants94 achieved the efficacy end point 9 and 18 at months when compared with the external placebo group; the study showed significant improvements in neuropathic impairment, disability aspects, nutritional status, gait speed, and quality of life; and half of the patients showed disease reversal relative to baseline in assessments of neuropathy and quality of life. The ongoing HELIOS-B study, a phase III, multicenter, double-blind, randomized controlled study (NCT04153149), aims to assess the incidence of cardiovascular events (CV) (hospitalization and urgent heart failure) and all-cause mortality in patients with ATTR-CA treated with vutrisiran.
Inotersen, a second-generation modified antisense oligonucleotide targeting transthyretin messenger RNA (mRNA), was approved by the US FDA for patients with ATTR-PN in October 2018. An OLE study, which was an international, double-blind, randomized, controlled study (NCT02175004) as a continuation of the NEURO-TTR study (NCT01737398), showed that inotersen treatment in patients with ATTR-PN delayed the deterioration of quality of life and disease progression.95 Luigetti et al96 confirmed this idea and suggested that earlier use of inotersen in patients would have been more beneficial. In a study by Dasgupta et al,97 33 patients treated with inotersen, at the 2-year time point, mean LV volume decreased by 8.4%, and the 6-minute walking test showed a 20.2 m increase in mean exercise tolerance. Further positive measures were observed after 3 years, not only was there no disease progression, but in some cases, the cardiac amyloid burden was reduced.
Eplontersen (ION-682884, AKCEA-TTR-LRx, IONIS-TTR-LRx) is a conjugate antisense oligonucleotide used to degrade TTR mRNA in the liver. Viney et al98 conducted a Phase I trial (NCT03728634) in healthy volunteers, which showed that AKCEA-TTR-LRx reduced TTR levels by approximately 90% after 4 injections of 90, 60, or 45 mg every 4 weeks. All doses were well-tolerated without serious adverse events. A population pharmacokinetic/pharmacodynamic model of eplontersen by Diep et al99 predicted the optimal dose for clinically significant and sustained TTR reduction to be 45 mg per 4 weeks. This provides a basis for dosing selection in future phase III clinical trials. A Phase III, multicenter, international study (NCT04136184) of ION-682884 for TTR neurotype is currently ongoing. In addition, in a global randomized controlled study100 (NCT04136171), 1438 patients will be 1:1 randomized to receive IONIS-TTR-LRx or placebo to observe recurrent cardiovascular clinical events and cardiovascular mortality after 140 weeks of IONIS-TTR-LRx treatment.
IONIS-TTR (ISIS 420915) is a second-generation antisense oligonucleotide with a 2-methoxyethyl modification. It targets the messenger RNA (mRNA) of TTR, which is complementary to the 30 untranslated regions of the mRNA and is bound by Watson and Crick base pairing. The binding leads to RNaseH1-mediated degradation of TTR mRNA, preventing101 the production of TTR protein. In the Benson et al study,101 15 patients who used IONIS-TTR, did not show ATTR-CA progression 1 year after treatment, which specifically inhibits TTR liver synthesis.
Gene-Editing TechniquesCRISPR/Cas9-mediated genome editing presents a significant challenge for new drug therapies in clinical practice. It has already demonstrated early success in clinical trials, where knocking down TTR expression in liver cells can address diseases caused by mutant or wild-type proteins.102 CRISPR-Cas9 genome editing with end-joining to specific TTR target sites induces DNA structural variations with low risk.103 Finn et al104 showed in a mouse trial that a lipid nanoparticle (LNP), a mediated delivery system in which CRISPR/Cas9 editing resulted in more than 97% knockdown of TTR protein in the mouse liver, which can be sustained for at least 12 months. In a small group of patients with ATTRv-PN (NCT04601051). Gillmore et al103 found that a single dose of NTLA-2001 (an in vivo gene-editing therapy) led to a continued reduction in serum TTR protein concentration by targeting TTR knockout. On day 28, the serum TTR protein concentration decreased by 87% and 52% in the 2 dose groups receiving 0.3 mg/kg and 0.1 mg/kg, respectively, and was associated with only minor adverse events. The safe and effective application of CRISPR/Cas9, as the latest advanced treatment in biotechnology, improves the prospect of the clinical application of gene editing.105,106
Amyloid Fiber Degraders (Monoclonal Antibodies)NNC6019-0001 is a humanized monoclonal antibody that targets a unique epitope exposed only to misfolded monomers and aggregated forms of TTR through antipt-mediated phagocytosis. A Phase III, randomized controlled trial (NCT05442047) recruited 99 patients with ATTR-CA to evaluate the effects of 100 mg/kg and 30 mg/kg of NNC6019-0001 on patient cardiac function end points and biomarkers, as well as pharmacokinetics, tolerability, and safety. This trial is currently ongoing.
NI301A (NI006) is a recombinant human monoclonal immunoglobulin that removes patient-derived TTR deposition by macrophages and removes TTR fibrils and deposition in mice in a time- and dose-dependent manner.31 A Phase I clinical trial (NCT04360434) for patients with ATTR-CA is currently ongoing.107
PRX004 is an intravenous humanized monoclonal antibody against TTR that promotes clearance of insoluble amyloid fibrils through antibody-mediated phagocytosis and inhibition of amyloid fibrillary formation.
Comments (0)