
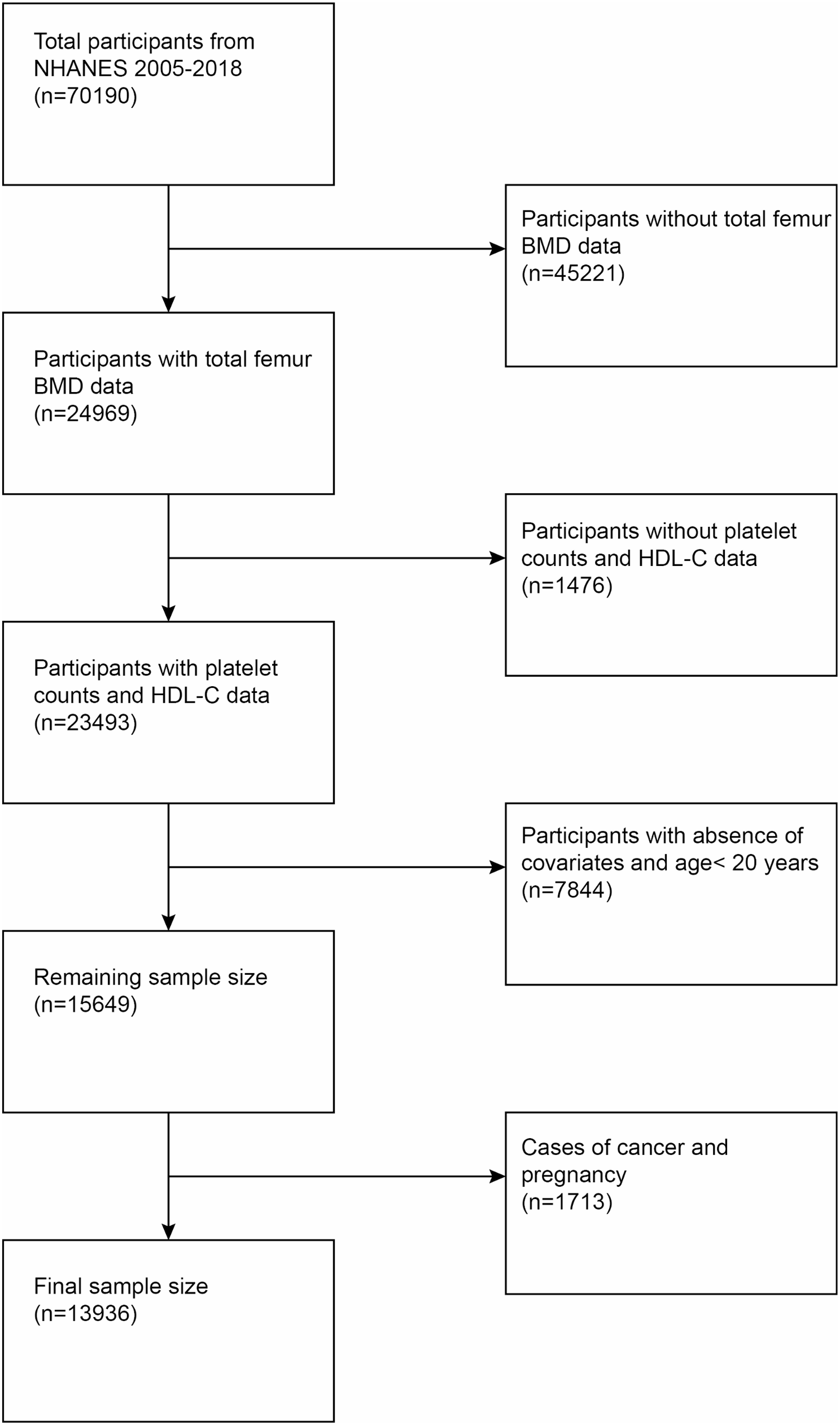
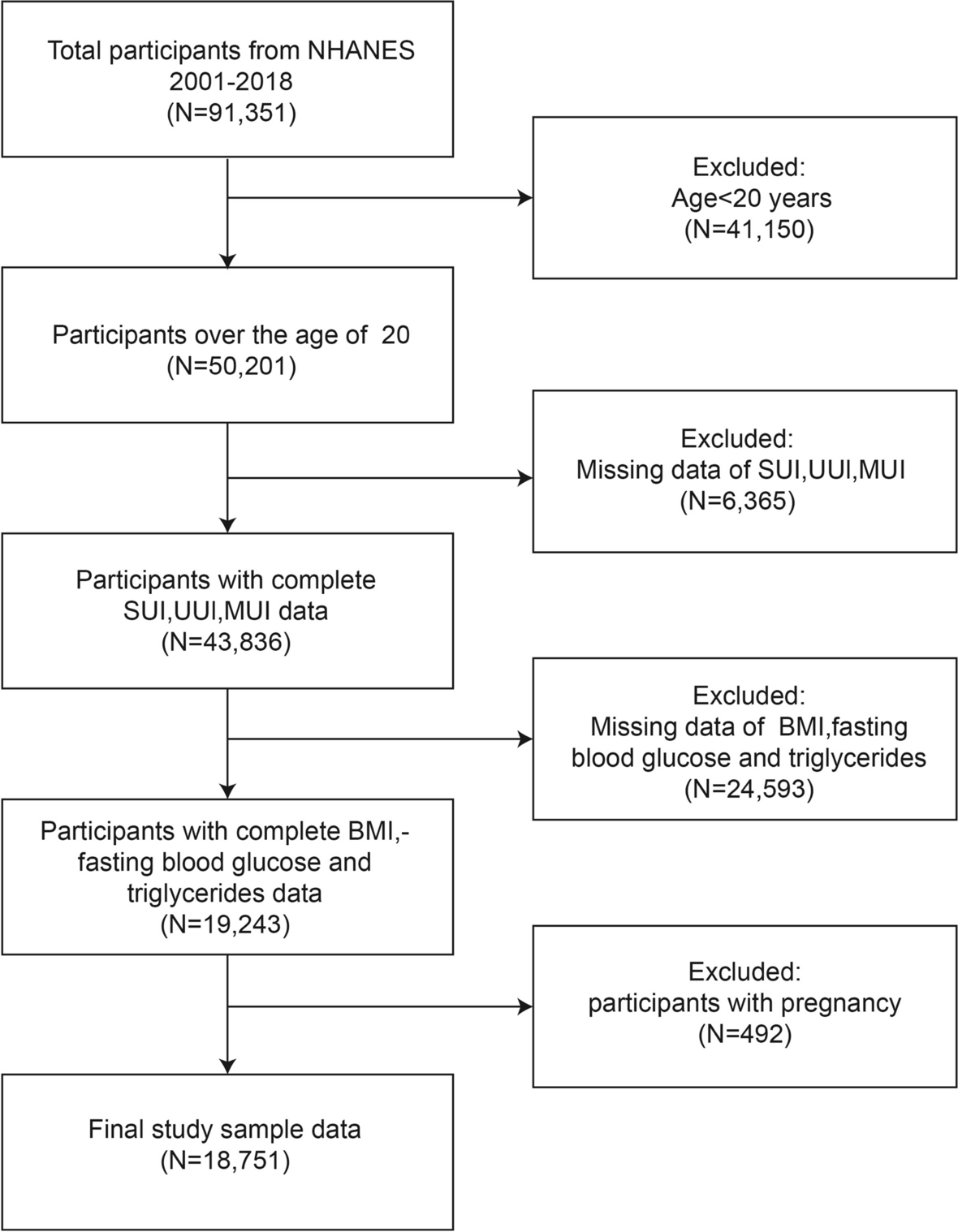
Remember me




According to the early study, the RA-30 group (30 mg/kg RA) showed alleviation of NASH signs, which was similar to that exhibited by the positive control GS-0976 group [16]. Therefore, primary hepatocytes isolated from the control group, HFD-induced NASH group, and RA-30 (30 mg/kg RA) group were used for transcriptomics and proteomics analyses.
The yields of primary hepatocytes in the control, NASH, and RA groups were approximately 4.5 × 107, 1 × 107, and 1.44 × 107 cells/per mouse, respectively (Supplementary Table 1). The quantity was sufficiently high for conducting downstream transcriptome or proteome analysis.
On average, 11.75 Gb of sequenced nucleotides was obtained from the primary hepatocytes of each sample, with a genome mapping rate of 97.66% (Supplementary Table 2).
Of these, 3164 genes showed significantly changes between the control and NASH groups (1678 and 1486 were upregulated and downregulated, respectively) (Fig. 2A). A significant difference in 1797 genes was observed between the NASH and RA groups (340 and 1457 genes were upregulated and downregulated, respectively) (Fig. 2B). Overall, 936 intersecting DEGs were probably target genes involved in NASH pathogenesis and RA treatment (Fig. 2C and Supplementary Table 3).
Fig. 2
An overview of transcriptome and functional enrichment analysis of DEGs. A-B Global gene expression changes of M vs. C (A) and RA vs. M (B) are plotted as volcano plots. DEGs in red are upregulated, DEGs in green are downregulated, and non-DEGs are highlighted in gray. C Venn diagram of intersecting DEGs of M vs. C and RA vs. M. D-E Top 10 categories for GO biological processes (D) and KEGG pathways (E) of DEGs depicted by bubble diagrams. The color indicates the Q value, and the size indicates the gene number of each pathway. C: Control group. M: NASH group. RA: RA-30 group. N = 10 per group
For functional classifications, GO analysis was performed for the 936 intersecting DEGs. The top 10 GO terms were focused on, which mainly included the immune process and the inflammation response (Fig. 2D). The physiological roles of these DEGs were identified based on the KEGG analysis. (Fig. 2E). Among the top 10 pathways, 6 pathways (namely, Fc gamma R-mediated phagocytosis, phagosome, antigen processing and presentation, and chemokine, Toll-like receptor, and C-type lectin receptor signaling pathways) were related to the immune system. Moreover, the tumor necrosis factor (TNF) signaling pathway can trigger multiple intracellular signals affecting inflammation and immunity. These results are consistent with those of the GO analysis.
According to the comprehensive GO and KEGG analyses, RA might exert anti-inflammatory effects by regulating the inflammation-inducing immune response.
Network diagram analysis of the transcriptomeThe top 10 pathways with the most genes were categorized into two clusters through KEGG enrichment analysis (Fig. 3A).
Fig. 3
Network analysis of transcriptomics. A Pathway relation network of the top 10 KEGG pathways in DEGs. B The PPI network for DEGs in Cluster 1. C PPI hub genes ranked by degree in Cytoscape (ver.3.9.0). Node degree is represented by the redness of the nodes
Cluster 1 primarily comprised the immune system (antigen processing and presentation, C-type lectin receptor, and chemokine signaling pathway), metabolic pathway (oxidative phosphorylation), and TNF signaling pathway related to inflammation and immunity. Cluster 2 included the ribosome, which may contribute to NASH development, but the mechanism of action of the ribosome in NASH has still remained unclear [28,29,30].
Both KEGG and GO analyses indicated that the regulation of the immune/inflammatory response was largely enriched. Therefore, Cluster 1 was selected as the core cluster for identifying hub genes. The STRING tool was utilized for establishing the PPI network. This network consisted of 125 nodes interacting with each other through 271 edges (Fig. 3B). The DEGs were enriched in oxidative phosphorylation and formed an independent cluster. Oxidative phosphorylation is associated with oxidative stress-induced liver injury [31]. However, RA had no obvious effect on hepatic injury and could not reverse the decrease in oxidative phosphorylation in the NASH group (Supplementary Fig. 3) [16]. Therefore, the other cluster containing more genes was selected and imported into Cytoscape. The top 10 hub genes were identified based on the Degree in cytoHubba: Rous sarcoma oncogene (SRC), TLR4, Rac family small GTPase 2 (RAC2), TNF, spleen tyrosine kinase (SYK), phospholipase C gamma 2 (PLCG2), histocompatibility 2 (H2-Eb1), vascular cell adhesion molecule 1 (VCAM1), vav 1 oncogene (VAV1), and C-X-C motif chemokine ligand 10 (CXCL10) (Fig. 3C and Supplementary Table 4). Among these hub genes, TLR4 represents the intersection of metabolism and immunity, thereby playing a vital role in HFD-induced inflammation. TLR4 also regulates the expression of the inflammatory cytokine TNFα through the classical pathway [11, 32, 33]. The PLCG2-IP3-Ca2+ cascade activates TLR4 translocation, and TLR4 mediates the expression of IRF3 regulatory genes with SYK [34,35,36]. In addition, CXCL10 plays a crucial role in recruiting macrophages and is associated with the induction of proinflammatory cytokines (TNFα, IL-1β) [37].
Hence, hub genes were enriched in TLR4-mediated inflammation, a process that generally contributes to fatty liver disease and regulates proinflammatory cytokines expression.
Effects of RA on NASH at the proteome levelIn total, 15,958 peptides and 3493 proteins were identified for the DIA proteomic analysis (Supplementary Table 5). In total, 1118 proteins were differentially expressed in the NASH versus control comparison. Of these, 958 were upregulated and 160 were downregulated. In comparison with the NASH group, the RA group exhibited significant changes in 766 proteins (111 and 655 were upregulated and downregulated, respectively). Most proteins exhibited a smaller fold change in expression between the RA and control groups, indicating that the overall protein level tended to be normal after RA treatment (Fig. 4A). In total, 514 intersecting proteins were selected. (Fig. 4B and Supplementary Table 6).
Fig. 4
An overview of the proteomics of liver samples from HFD-fed mice and functional enrichment analysis of DEPs. A Heatmap displaying protein expression changes. Downregulated DEPs are shown in blue, and upregulated DEPs are shown in red. B Venn diagram of DEPs. C-D Top 10 categories for GO biological processes (C) and KEGG pathways (D) of DEGs depicted by bubble diagrams. The colour indicates the Q value, and the size indicates the gene number of each pathway. C: Control group. M: NASH group. RA: RA-30 group. N = 10 per group
The selected DEPs were assigned to GO categories to determine the biological processes in which they were involved. These DEPs were mainly associated with RNA processing and splicing, protein localization, amino acid metabolism, carbohydrate metabolism, and oxidation–reduction (Fig. 4C). The top 10 enriched pathways according to the KEGG enrichment study included carbohydrate metabolism, amino acid biosynthesis, RNA splicing and transport, and ferroptosis (Fig. 4D). The results of KEGG and GO analyses were consistent in terms of carbohydrate metabolism, amino acid metabolism, and RNA splicing. These pathways might be the key pathways for RA in hepatocytes.
Network diagram analysis of proteomicsUsing the same method as previously, the top 10 pathways were selected to build the KEGG network based on the KEGG enrichment analysis. Two clusters were created from these paths (Fig. 5A). One cluster was mainly related to metabolic pathways and biosynthesis of amino acids and antibiotics, while the other cluster was related to RNA transport and the spliceosome.
Fig. 5
Top 10 hub genes of proteomics revealed by network analysis. A The pathway relation network of the top 10 KEGG pathways in DEPs. B The PPI network for DEPs in Cluster 1. C PPI hub genes ranked by degree in Cytoscape (ver.3.9.0). Node degree is represented by the redness of the nodes
Due to the fact that the complicated interaction of metabolic pathways in the liver is the basis of NASH pathogenesis, the metabolism-related module (Cluster 1) was selected for the protein interaction analysis. Protein interactions were analyzed using the STRING database. These interactions indicated that 88 DEPs were functionally linked with each other through 77 edges (Fig. 5B). Based on the degree in Cytoscape software, 10 hub proteins were selected. These proteins revealed the involvement of proteins associated with glucose metabolism: glucose-6-phosphate isomerase 1 (GPI1), triosephosphate isomerase 1 (TPI1), phosphoglucomutase 2 (PGM2), transketolase (TKT), aldolase B (ALDOB), HK2, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), phosphoglycerate kinase 1 (PGK1), glutamic-oxaloacetic transaminase 1 (GOT1), and fructosebisphosphatase 1 (FBP1). These findings indicate that RA might strongly impact glucose metabolism (Fig. 5C and Supplementary Table 7).
Integrative pathway based on proteome and transcriptome data analysesTo compare direction-related changes in mRNAs and proteins, 1576 proteins were identified that had corresponding mRNA data (FDR ≤ 0.001) in the NASH versus RA comparison, and their differences (fold change ≥ 1.5) were classified according to the direction of change (Fig. 6A and Supplementary Table 8). (Fig. 6A).
Fig. 6
Integrative analysis based on proteome and transcriptome data. A Comparison of the expression changes in mRNA and protein. Blue: decreased mRNA and increased protein levels (n = 9); green: decreased mRNA and protein levels (n = 152); red: increased mRNA and protein levels (n = 9); yellow: increased mRNA and decreased protein levels (n = 4). B-C Top 10 GO biological process categories (B) and top 10 KEGG pathways (C) of DEPs/DEGs (green group), as depicted by bubble diagrams. The colour indicates the Q value, and the size indicates the gene number of each pathway. C: Control group. M: NASH group. RA: RA-30 group. N = 10 per group
Functional enrichment analysis was applied to 154 genes concordant with decreasing mRNA and protein levels (green group). The GO analysis revealed that the most enriched processes were the oxidation–reduction process (response to oxidative stress), carbohydrate metabolism (ethanol catabolic process and glycolytic process), immune process (antigen processing and presentation, cellular response to interferon-γ), and protein transport and folding (Fig. 6B). Notably, according to the KEGG pathway analysis of the green group (n = 152) (Fig. 6C), glycolysis/gluconeogenesis was the first among the top 10 pathways ranked by the Q value. This finding was highly consistent with the proteome results showing that RA could affect glucose metabolism.
The expression patterns of the remaining groups (red, blue, and yellow) were visualized through a heatmap (Supplementary Fig. 4). Among these, cytochrome P450 CYP4A14 and CYP4A10 contribute to fatty acid oxidation [38,39,40,41] and their mRNA and protein levels were both greatly increased in the NASH group, consistent with previous studies [16]. This result indicated that RA could improve the fatty acid oxidation capacity in mice with NASH. Moreover, the level of insulin-like growth factor binding protein 2 (IGFBP2) is correlated with hepatic steatosis inversely [42], and both its genes and proteins were elevated in the RA group in the present study.
Based on the protein and gene expression data, the GO and KEGG annotations and hub genes in the proteome and transcriptome analyses, and the correlation results, integrated pathway maps were constructed. Key genes in the TLR4/AP1 pathway and glycolysis were downregulated. After RA treatment, TLR4, MyD88, mitogen-activated protein kinase kinase kinase 8 (Map3k8, also named TPL2), mitogen-activated protein kinase 3 (Mapk3, also known as ERK), and AP1 subunits (c-Fos and c-Jun) were significantly downregulated (Fig. 7A and Supplementary Fig. 5). The key enzymes for glycolysis, that is, HK2, HK3, PFKL, PKM, and lactate dehydrogenase A (LDHA), were downregulated in the RA group versus the NASH group (Fig. 7B and Supplementary Fig. 6).
Fig. 7
Integrated KEGG pathway maps. DEGs and DEPs were mapped to the Toll-like receptor signaling pathway (A) and glycolysis/gluconeogenesis (B) (the change in mRNA and protein expression is expressed as log2 [fold change])
Therefore, the role of RA in improving NASH might be achieved through a decrease in glycolysis and the TLR4/AP1 pathway.
Verification of the effect of RA on the TLR4/AP1 pathwayFour key genes of the TLR4/AP1 signaling pathway were examined in primary hepatocytes: TLR4, MyD88, and AP1 subunits (c-Fos and c-Jun). The mRNA expression of MyD88, c-Fos, and c-Jun were markedly elevated in the NASH group relative to the control group (Fig. 8A). Downregulation of TLR4, MyD88, and AP1 subunits was observed in the RA group relative to the NASH group, which indicates that a crucial mechanism through which RA might suppress inflammation is by altering the mRNA expression of genes in the TLR4/AP1 signaling pathway in hepatocytes.
Fig. 8
Effect of RA on the TLR4/AP1 pathway. A-B mRNA expression of TLR4, MyD88, c-Jun, and c-Fos in primary hepatocytes (A) and in primary Kupffer cells (B). C-D The protein abundances (C) and protein expression (D) of TLR4, MyD88, c-Fos, phospho-c-Fos, c-Jun, and phospho-c-Jun in primary Kupffer cells. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the NASH group. C: Control group. M: NASH group. RA: RA-30 group. N = 10 per group
Most studies on TLR4 signaling have focused on nonparenchymal cells, such as Kupffer cells [43]. Therefore, the above-mentioned four key genes were examined in primary Kupffer cells.
In the HFD-induced NASH group, the relative MyD88, c-Fos, and c-Jun mRNA levels were markedly elevated by 38%, 82%, and 163%, respectively, compared with the control group. RA treatment reversed the increase in MyD88, c-Fos, and c-Jun mRNA levels and decreased TLR4 mRNA levels by 37% in comparison with those of the NASH group (Fig. 8B).
The RA-regulated TLR4/AP1 pathway was further explored by measuring the protein levels of TLR4, MyD88, c-Fos, phospho-c-Fos, c-Jun, and phospho-c-Jun. In comparison with the control group, a 34% increase was observed in the expression of TLR4 and MyD88 (P < 0.05), whereas a trend of increase in c-Jun and c-Fos expression was observed (Fig. 8C, D). After RA treatment, c-Fos, phospho-c-Jun, and phospho-c-Fos expression levels decreased by 14%, 23%, and 24% (P < 0.05), respectively, and TLR4 expression exhibited a decreasing trend. However, MyD88 and c-Jun expression remained unchanged. Because of the action of RA, TLR4 expression and AP1 phosphorylation decreased. Thus, RA could reduce inflammation in HFD-induced mice by modulating the TLR4/AP1 signaling pathway expression.
Verification of the effect of RA on glycolysisThe DEGs/DEPs identified through transcriptomic and proteomic analyses revealed that key glycolytic enzymes were significantly regulated in the liver (Fig. 7B). The expression of glycolysis-related genes (HK2,6-phosphofructo-2-kinase (PFKFB3), PKM2, PFKL, and enolase 1 (ENO1)) was relatively quantified through qPCR. The RA group exhibited significantly decreased mRNA levels of the aforementioned genes in comparison with the NASH group, with the mRNA levels of PFKFB3, PKM2, and PFKL distinctly increased by 452%, 15%, and 188%, respectively, in the NASH group after the mice were fed a HFD (Fig. 9A).
Fig. 9
Effect of RA on the key factors involved in glycolysis. A The mRNA expression levels of hepatocyte HK2, PFKFB3, PKM2, PFKL, ENO1, LDHA, ChREBP, and MLX. B-C The protein abundances (B) and protein expression (C) of hepatocyte HK2, PKM2, PFKL, and ChREBP. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the NASH group. C: Control group. M: NASH group. RA: RA-30 group. N = 10 per group
LDHA is predisposed to converting pyruvate into lactate, which is the final step in glycolysis [44]. LDHA mRNA expression exhibited a decreasing trend in the NASH group relative to the control group and was substantially decreased in the RA group by 23% relative to the NASH group (P < 0.01). These data suggested that RA exerts a suppressive effect on glycolysis and changes LDHA mRNA expression (Fig. 9).
Glycolysis regulates the transcription factor ChREBP, which interacts with Max-like protein (MLX), thereby affecting the expression of lipid synthesis genes [45]. In this study, the ChREBP and MLX mRNA expression were significantly increased by 275% and 103%, respectively, in the mice fed a HFD. In contrast to the NASH group, RA treatment reversed the enhanced MLX and decreased ChREBP mRNA levels by 37% (Fig. 9A).
The aforementioned results were verified through western blotting to confirm HK2, PKM2, PFKL, and ChREBP expression levels in mouse primary liver cells. HK2 protein levels were enhanced by 40% after the mice were fed a HFD, whereas PFKL, PKM2, and ChREBP protein levels exhibited an upwards trend. Following RA treatment, HK2, PKM2, PFKL, and ChREBP expression decreased to the normal physiological level (Fig. 9B-C). Consequently, RA inhibited glycolysis and ChREBP to restore inflammation and lipogenesis in mice with NASH.
Effect of RA on arsenic-induced glycolysis in vitroTo further confirm whether RA acts as an inhibitor of glycolysis, the effect of RA on arsenic-induced glycolysis in normal liver cells (L-02) was assessed according to published methods [46, 47].
NaAsO2 (0.2–12.8 μM) exhibited no conspicuous inhibitory effect on L-02 cell growth (Fig. 10A). Lactate production in these cells increased significantly after treatment with different NaAsO2 doses for 24 h (Fig. 10B).
Fig. 10
The effect of RA on glycolysis in NaAsO2-treated L-02 cells. A Viability of L-02 cells treated with various NaAsO2 doses for 24 h. B The release of lactate in L-02 cells treated with various NaAsO2 doses. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. 0 μM NaAsO2 treatment. C L-02 cells release lactate in response to NaAsO2 and RA. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. N = 3 per group
The extracellular lactate level was enhanced in the NaAsO2-treated group relative to the control group (P < 0.001) (Fig. 10C). RA itself had no effect on lactate production. RA concentrations of 10 and 30 μM were used in the study according to a previous publication [16]. Compared with the NaAsO2-treated group, lactate production in the RA-treated group decreased dose-dependently (P < 0.01) (Fig. 10C). These studies revealed that RA reduces NaAsO2-induced lactate production in L-02 cells.
Overall, RA decreased glycolysis to reduce lactate production, thereby alleviating inflammation and excessive lipogenesis.
Comments (0)