
Remember me


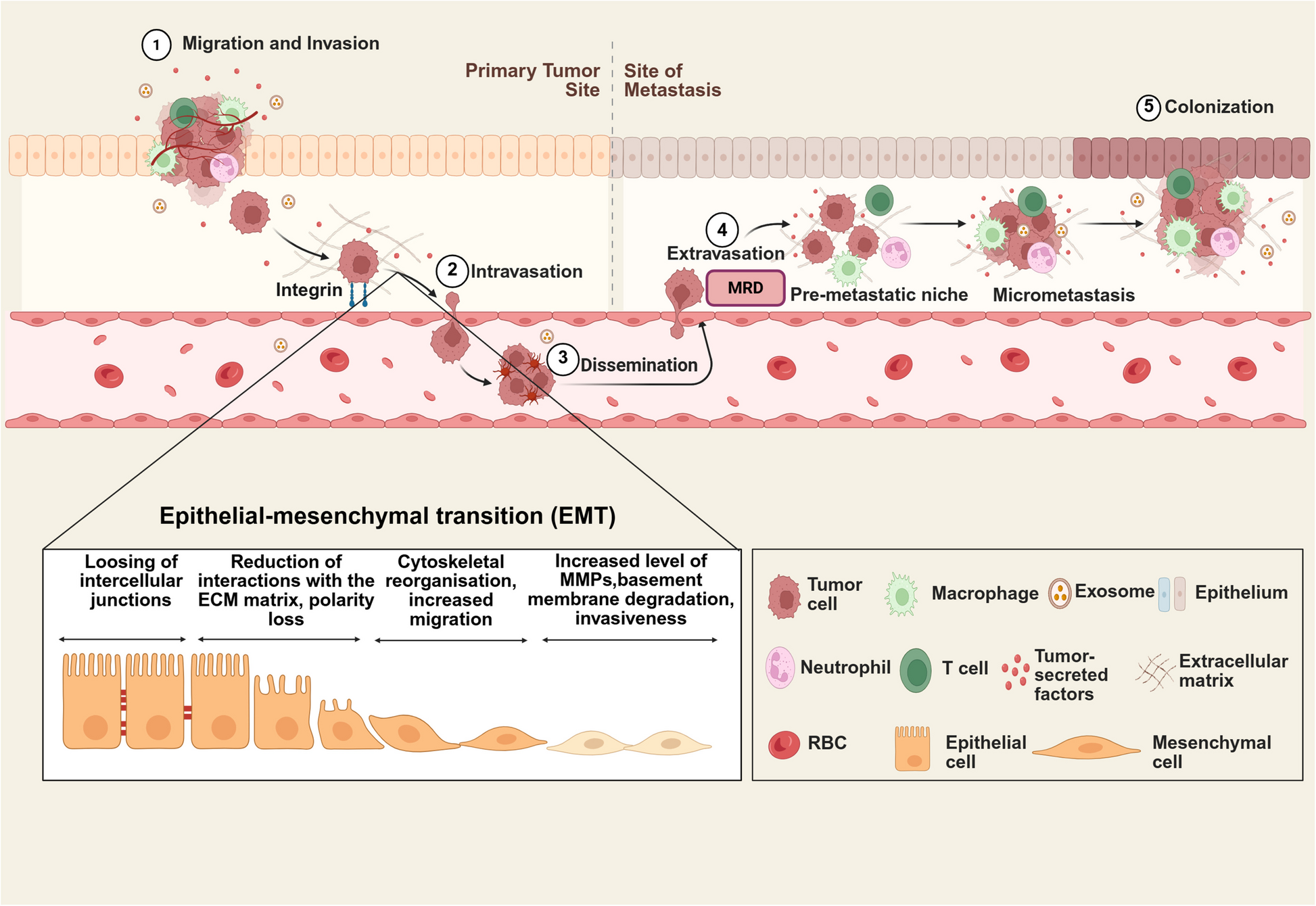
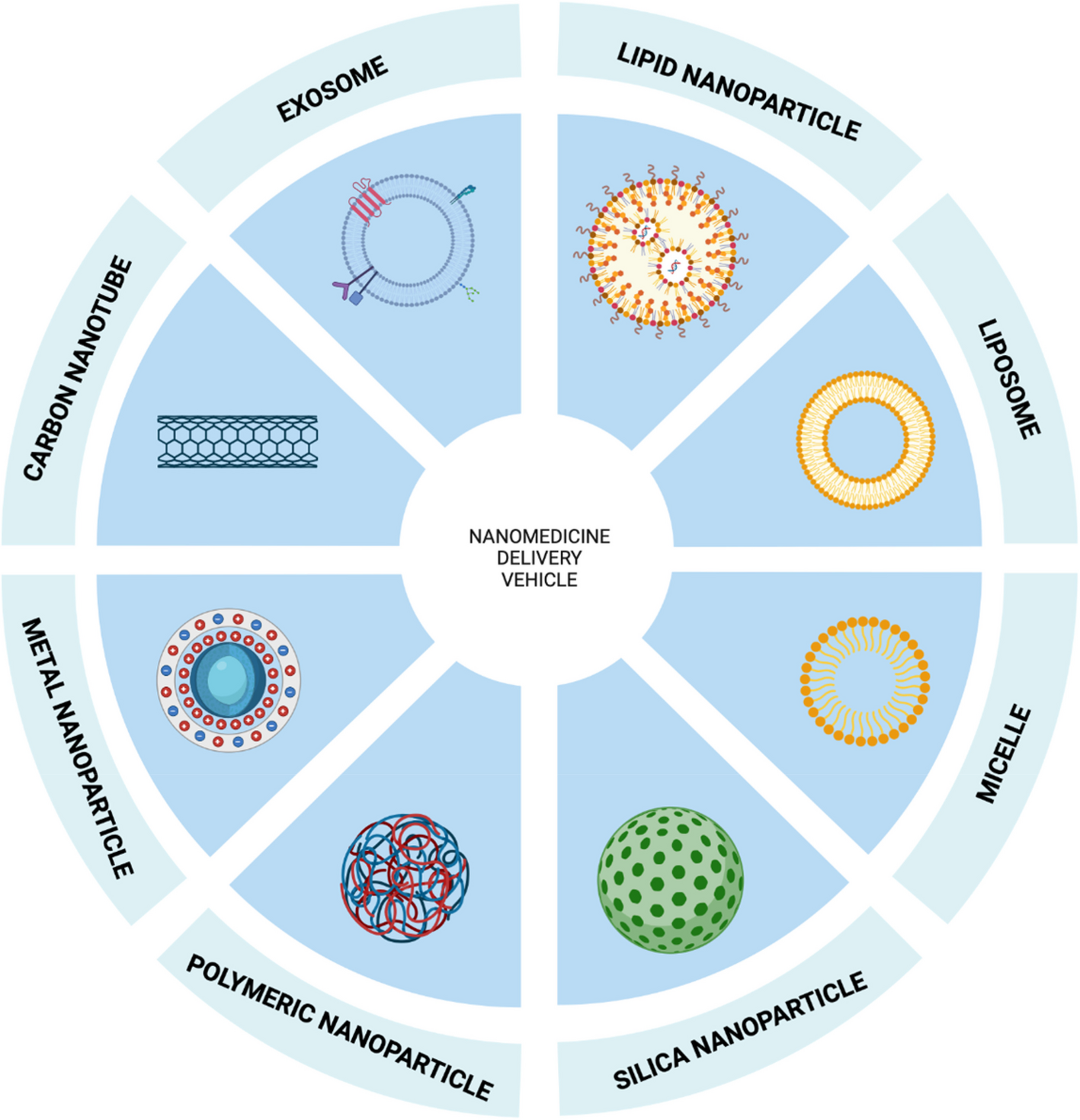
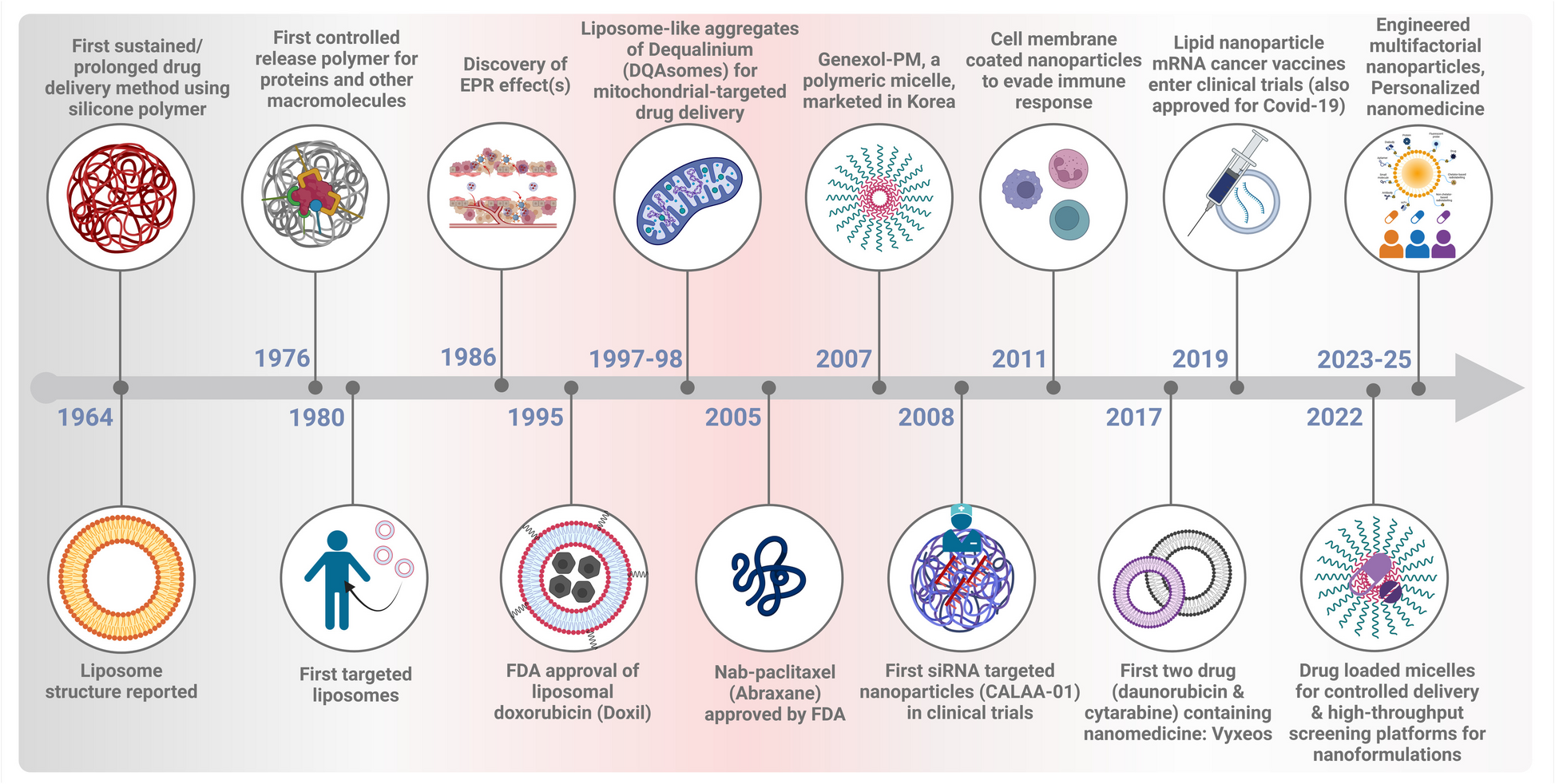
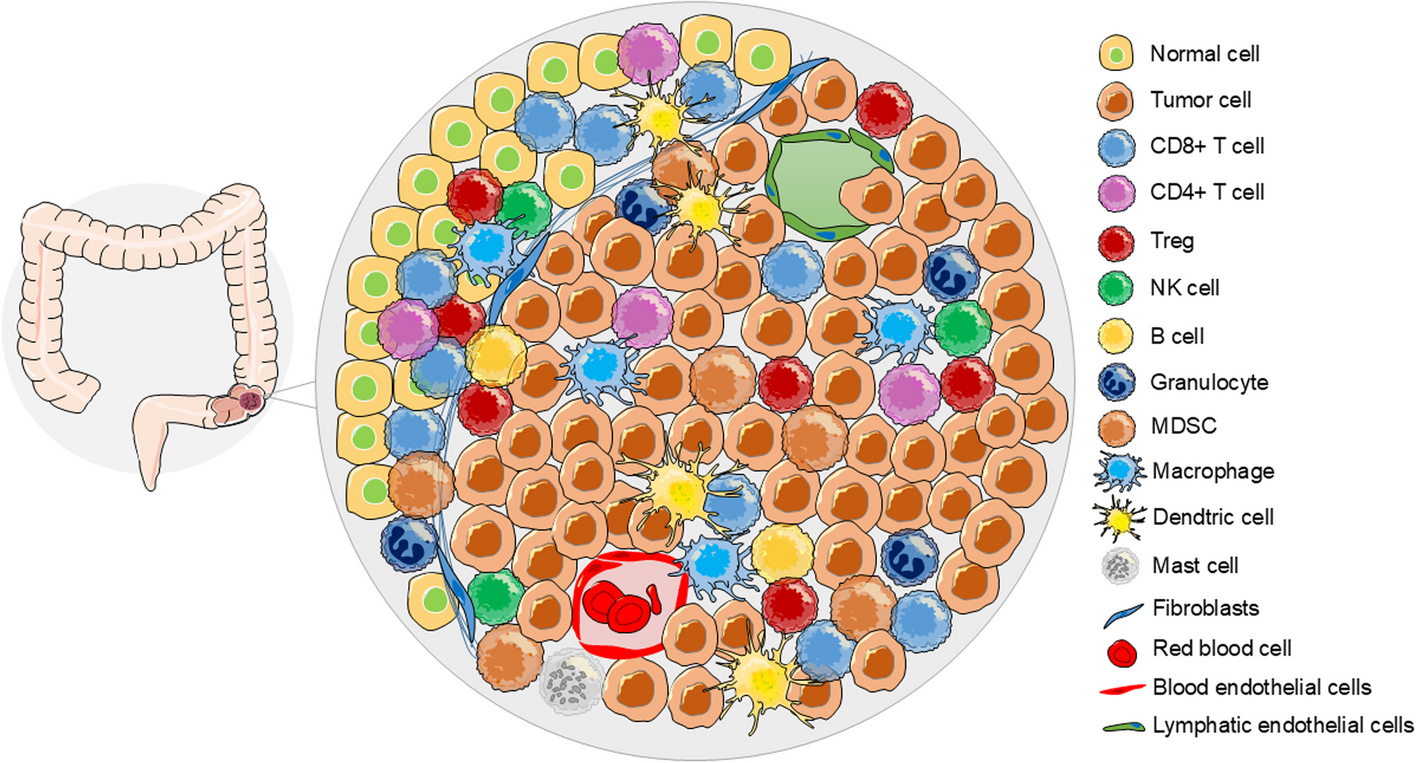
The study was conducted to investigate the association between 26 immune gene sets (listed in Table S1) [7] and immune checkpoint proteins expressed in HNSCC and their effect on patient survival. In the current study, we assessed these signatures in HNSCC, as 20 of the 26 gene sets were also found to be modulated in HNSCC [8]. In particular, in the present analysis we assessed the effect of the 26 immunosignature on both the programmed cell death protein ligand-1 (PD-L1) axis and the T-lymphocyte associated protein 4 (CTLA-4) axis, controlling for potential confounders and effect modifiers [9]. In the 520 participants of the TCGA the variables relating to the characteristics of the patients showed how adulthood, between 53 and 69 years, was the one with the highest prevalence with 48% of cases represented in that range as well as the male versus female gender with 74% of the cases considered. The distribution by tumor volume showed similar frequency between T2, T3 and T4 (29%, 26% and 35%, respectively), as well as the nodal status with 47% for N0 50% for N + . When we looked at the disease stage, stage IV represented half of the observations, while stage II and III were equally distributed around 20% each and stage I, instead, showed the smallest frequency with 4% of the observations. As expected, HPV negative cases were the vast majority of cases compared to positive cases (81% versus 19%) as well as alcohol consumers (67%) versus no-alcohol consumers. Active smokers showed a slightly higher frequency than non-smokers (34% versus 22%) and ex-smokers who quitted more recently (less than 15 years) showed a higher frequency of cancers than those who stopped smoking for a longer period of time (greater than 15 years). As regards the mutational characteristics, lesions with TP53 mutations were more frequent than those without mutation (70%) while the mutated forms of CDKN2A, FAT1 and PIK3CA showed a lower frequency of mutated forms than those non-mutated with 78% of non-mutated cases for CDKN2A, FAT1 and 82% for PIK3CA (Table S2). The 26 immune gene expression signatures, the related analysis of the genes that comprise these signatures, and the analysis of the 15 immune cell types, are described in Table S1.
To test the hypothesis that TP53 gene status and co-driver mutations could be combined to prognostically stratify HNSCC patients and potentially influence the response to ICI, we used a step-by-step approach depicted in the Fig. 1A flowchart.
Fig. 1
A Workflow of the main analyses. B Forest plot representing the association of average expression of 125 genes included in the 26 immune gene sets and the clinical variables in 520 HNSCC patients from TCGA. Results of the linear regressions are shown as Odds Ratio with confidence intervals at 95%. C-D Kaplan–Meier curves of HNSCC patients from TCGA cohort with high or low Immune Scores evaluated for overall survival and progression free survival (panels C and D, respectively). Differences between curves were evaluated by logrank test. Hazard ratios with 95% confidence intervals were assessed by Cox Hazard regression models. Immune Scores were evaluated as the positive and negative z-scores of the average expression of the 125 genes composing the immune gene sets. E Overall Survival in a cohort of 108 HPV-negative HNSCC patients (Huang et al.). Patients were divided based on high and low levels of the Immune Score. Differences between curves were evaluated by log-rank test. F Distributions of the gene signature composed by the average expression of 125 genes of the immune gene sets by TP53 mutation and TP53 mutation carried on other mutations among FAT1, CDKN2A and PIK3CA in HNSCC patients (106 WT, 171 TP53 and 189 TP53 + mutX). P-values were evaluated by KruskalWallis test. G Distributions of the PDL1 protein among different mutational status subgroups from a set of 339 HNSCC patients evaluated by reverse phase protein array (RPPA) in the TCGA cohort. H Gene set enrichment analysis of co-mutated patients versus TP53 mutated patients in the TCGA HNSCC cohort. The size of the circles indicates the percentage of genes included in the pathway. Pathways are sorted by False Discovery Rate and normalized enrichment score (NES). The PI3K pathway and the MYC pathway activity were highlighted. For the analysis, we used the GSEA 4.2 software (https://www.gsea-msigdb.org/gsea/index.jsp) run in pre-ranked mode with HALLMARK pathways. I Pearson’s correlation between the mean expression of 26 immune gene sets (upper panel) and PD-L1 expression values (bottom panel) with the levels of expression of a 22 genes signature MYC dependent (Ganci et al.) in HNSCC patients from TCGA. A Multivariate regression models were built to adjust the differences of the genes between patients with high and low MYC signature. The models include T status, TP53 mutation, gender, smoking status and, HPV status. High and low expression of the MYC signature were evaluated by positive and negative z-scores of the mean gene expression, respectively. J qRT-PCR analysis of PD-L1 in Cal27, FaDu and Detroit 562 cell lines. Statistics (t-test): * p < 0.01, ** p < 0.005. K Flow cytometry analysis of PD-L1 surface expression in cell lines. Representative cell lines (color-coded) were harvested from their cultures and stained with CD274-PE mAb or control Ig for 30 min at 4 °C. Surface expression was assessed on single, live cells on the Attune NxT cytometer. Mean fluorescence intensity is shown. The staggered plot depicts cell line expression according their mutational status
Step 1 of the flowchart includes the computational analysis of the clinical genomics data from TCGA. Phase (A) describes the analysis of 26 immune gene expression signatures as prognosis predictors within the TCGA cohort of 520 HNSCC patients. A global immune score (IS) for the 26 gene sets considered was obtained as the z-scores of the average expression of the genes included in the gene sets. Phase (B) describes the analysis of the immunosignature prediction performance by HNSCC TCGA subgroups, such as TP53 wild type (TP53-WT) versus mutated (TP53-mut), alone (i) and with other co-mutated genes (ii). Phase (C) describes the analysis of association between IS and a 22 MYC-related genes expression. In our previous work we identified a 22-gene MYC-related signature in HNSCC cancer, which is specifically activated by TP53 mutations with a gain of function activity, and could therefore serve as a proxy for such mutations [10]. In Step 2, we evaluated the impact of aneuploidy in the immune signatures prediction and we performed a cell type enrichment among subgroup of patients with different mutational status. Finally, two validation cohorts of 102 and 139 HNSCC patients treated with PD-L1 inhibitor from the GEO database and MSKCC dataset were analyzed.
STEP 1 - Phase A: Regression and survival analysisIn Fig. 1, panel B, we aimed to identify subgroups of HNSCC patients with a significant difference of IS levels. The first set of results are represented by the forest plot with Odds ratio with 95% CI of demographic and prognostic predictors of the immune signature expression by using regression models in the HNSCC dataset from TCGA. All the analyses were performed at univariate level considering the effect of only one independent variable (demographic or prognostic variables) on the signature (dependent variable). We observed that only the HPV status, with HPV negative versus the positive lesions, lymphnode status (N0 vs N +) and TP53-mut plus additional mutations versus TP53-mut-only lesions, were statistically associated with a higher immune signature expression. Details of regression analyses on clinical factors for each immune gene set are shown in S-Fig. 1 and S-Fig. 2. Notably, building a multivariable regression model, TP53 mutational status resulted in the main clinical factor significantly associated with the immune signature (Table S3). While the relationship between TP53-mut patients and HPV-negative individuals is established, our observations reveal comparable percentages of HPV-negative patients in TP53-mut and TP53-mut plus additional mutations groups (91% and 93%, respectively). Taken together, these results suggest that the association between the immune gene set and mutational status, as defined by our categorization, remains unaffected by HPV status. To provide clinical meaning to the described results, in Fig. 1, panel C and D, we reported overall-survival (OS) and progression-free-survival (PFS) curves assessed in the TCGA cohort of 520 patients. In that cohort, high immune signature expression was significantly associated with both overall and progression-free survival. In Fig. 1, panel E, the prognostic value of the immune signature was further validated in the HNSCC CPTAC cohort, as described by Huang et al. [11]. The cohort consisted of 108 HPV-negative patients. This validation study aimed to assess the potential of the immune signature as a prognostic marker specifically for this subset of HNSCC patients confirmed the findings reported for TGCA (Fig. 1E). Additionally, in Supplementary Figure S-Fig. 3, we presented the prognosis of HNSCC patients in the TCGA cohort. Panel A excluded patients with pM1, while panel B excluded HPV-positive patients. This analysis provided insights into the different prognoses observed within the TCGA cohort based on these specific criteria.
Fig. 2
A The Spearman’s correlation coefficient reveals a negative association between aneuploidy score and immune signature. B Spearman's correlation of PDL1 with aneuploidy scores in TCGA HNSCC patients. C Spearman's correlation of the 22-gene MYC signature (Ganci et al.). D Distributions of the aneuploidy scores between TP53 mutated patients, WT patients and co-mutated patients. Co-mutated patients show lower aneuploidy than TP53 mutated patients. Statistical significance was evaluated by Wilcoxon test. E Forest plot and multivariate regression model to assess the weights in the immune gene sets prediction of the aneuploidy score and the TP53 co-mutational status. The variables resulted to be independent predictors of the immune signature. F Cell types enrichment analysis by comparing 64 cell type signatures in subgroups of HNSCC patients with TP53 mutation, TP53 mutation with other mutations and wild type patients. Heatmap representing the normalized average scores obtained from Xcell software, reflecting the cell type abundance of the most significant modulated cell types among the three subgroups. The statistical significance (p < 0.05) was assessed by KruskalWallis test. G Overall survival (left panel) and Progression free survival (right panel) of 102 patients treated with PDL1 inhibitors from GEO database (GSE159067). Patients were split basing on the Immune Score. The high\low levels of Immune Score were obtained considering the positive and negative z-scores of the average expression of the 26 immune gene sets, respectively. Differences between curves were evaluated by logrank test. The multivariate Cox Hazard regression analysis was adjusted for gender and HPV status. H Average expression of the 26 immune gene sets and MYC signature distribution in 102 patients treated with PDL1 inhibitors (GSE159067, left and right panel, respectively). The immune gene sets expression was evaluated in patients with complete or partial response and patients with stable disease or progression disease after treatment (Fig. 2H, left panel). The MYC signature expression was evaluated according to the phenotype classification (“COLD” and “HOT” patients) obtained from Foy JP and colleagues (Fig. 2H, right panel). Differences were evaluated by Wilcoxon test. I The overall survival of 139 HNSCC patients in Samstein's cohort (MSKCC) who underwent ICI treatment was analyzed based on their mutational status. P-values were assessed using the logrank test
STEP 1 - Phase B: Immunosignature in TP53 mutated (i) and TP53 co-mutated patients (ii)Because the mutational TP53 status was so important, we assessed the expression distribution of the immune signature by three groups of patients with TP53-WT status, TP53-mut status, and TP53-mut in combination with one of the other three most frequent mutations observed in HNSCC cancer patients (FAT1, CDKN2A, and PIK3CA genes), hereinafter denoted as TP53-mut+. TP53-WT patients were characterized by a higher IS expression level compared with TP53-mut and TP53-mut+ patients (Fig. 1F). We considered gene mutation regardless of mutation for statistical power considerations. However, most mutations are missense as described in the S-Fig. 4. Surprisingly, TP53-mut+ patients had a significantly higher IS score in comparison to the TP53-mut patients (Tukey’s post-hoc test, p < 0.05) though significantly lower than WT patients (Tukey’s post-hoc test, p < 0.01). Congruently, PDL1 protein expression was significantly higher in TP53-WT than in TP53-mut patients. The presence of a co-mutation over that on TP53 exhbited PDL1 protein expression comparable to TP53-WT patients (Fig. 1G). These findings were also found analyzing the HNSCC CPTAC cohort (S-Fig. 5B, C, and D, respectively), thereby providing further robustness to the findings reported for TGCA analysis. To ensure that the differential expression of the immune signature was not influenced by other genomic alterations, we conducted a comparative analysis using copy number alterations instead of co-mutations. Interestingly, we found no significant variation in the immune signature between patients with mutated TP53 and those with TP53 mutations along with other genomic alterations (S-Fig. 5A). This might suggest that the observed differences in the immune signature were specifically associated with TP53 mutations in presence of other co-mutations (TP53-mut+).
In Fig. 1H, we performed a gene set enrichment analysis (GSEA) using a ranking list of all genes, comparing their expression between co-mutated and TP53 mutated HNSCC patients. The majority of enriched pathways were related to the immune system, thus reinforcing the association between co-mutations and immune-related processes. Notably, we observed enrichment in MYC-related and PI3K/mTOR pathways among those enriched in the GSEA (Fig. 1H).
In TCGA HNSCC, we observed a negative correlation between the TP53 mutated-dependent MYC signature (from Ganci et al. [10]) and the immune signature (Fig. 1I). Furthermore, in S-Fig. 5E, we found a correlation between the gene PDL1 and the TP53 mutated-dependent MYC signature. These findings suggest the existence of a potential regulatory relationship between TP53, MYC, and immune-related pathways.
In addition, we found that PDL1 mRNA expression is higher in HNSCC cell lines carrying TP53 and additional co-mutations, such as Detroit-562 (TP53\CDKN2A\PIK3CA) and FaDu (TP53\CDKN2A\FAT1) compared to CAL27 carrying TP53\CDKN2A mutations (Fig. 1J). These findings paired with increased PDL1 surface staining in Detroit-562 and FaDu when compared to CAL27 HNSCC cell lines (Fig. 1K).
STEP 1 - Phase C: Association of a TP53 mutated-dependent MYC signature with the immune gene setsTo further detail the functional link between TP53 gene mutations with gain of function activity and immune signature, we assessed the role of the TP53 mutated-dependent MYC signature identified in our previous work [10]. In Fig. 1I, we assessed the correlation of the expression of the immune gene sets and PDL1 (S-Fig. 5E) in TCGA patients with high or low expression of MYC-related signature. A comparison of the same subgroups of patients was also conducted for CTLA4 in TCGA and for the immune gene sets in another HNSCC cohort from GEO (GSE195832). Again, lower expression level of the TP53 mutated-dependent MYC signature was significantly associated with higher levels of the IS score, PDL1 and CTLA4 (S-Fig. 6A, B, C and D). Furthermore, in TCGA cohort we had sufficient clinical information and sample size to adjust those modulations for potential confounding factors. The multivariate models reported at the bottom of panels A, B and C confirmed that those associations, between genes or IS and the MYC-dependent signature, were independent from other clinical factors. Interestingly, c-MYC protein expression was significantly higher in TP53-mut than in TP53-WT patients (S-Fig. 6E). In NCI-60 cell lines, indeed, we found that WT-TP53 HNSCC cell lines exhibit higher expression of CTLA4 and PDL1 when compared to cell lines harbouring TP53 mutations (S-Fig. 7A). We also observed that depletion of either mutant p53 protein or its co-factor YAP released PDL1 expression in HNSCC cell lines (S-Fig. 7B).
Enhanced expression of PDL1 was also obtained in CAL-27 and Detroit-562 after treatment with alpelisib, a selective inhibitor of p110α-subunit of PI3K (S-Fig. 7C).
We have previously identified that mutant p53 and YAP proteins favour c-Myc stability and its transcriptional activity in HNSCC cell lines [10]. In that context the use of alpelisib has been found to partially impair this pro tumorigenic axis [10].
To validate these results, we performed qRT-PCR analysis of PD-L1 (S-Fig. 7D) and CTLA4 (S-Fig. 7E) in Cal27 cells, a head and neck cancer cell line carrying a TP53 mutation, treated with JQ-1. The latter is a small-molecule that inhibits the activity of the BET family proteins by masking their bromodomain acetyl-lysine-binding pockets [12]. JQ-1 has been demonstrated to act as an antineoplastic agent by mainly inhibiting c-MYC functions. Both genes showed increased expression after treatment when compared to their controls, strengthening the potential role of MYC in this immunogenic context.
STEP 2 - Phase I: Analysis of aneuploidy and cell type enrichmentTo study the potential cause of the difference in IS expression between TP53-mut and TP53-mut + HNSCC patients, we considered the aneuploidy score of TCGA HNSCC patients. In general, aneuploidy is strongly associated with TP53 mutations, and is negatively correlated to several immune signatures across various cancers [13]. In line with previous evidence, we observed the negative correlation between aneuploidy scores and IS expression (Fig. 2A) and PDL1 expression (Fig. 2B). Additionally, we observed that higher levels of aneuploidy scores are associated with an increase in the MYC-dependent signature (Fig. 2C). As expected, aneuploidy levels were significantly higher in the TP53-mut and TP53-mut + patients (Fig. 2D). Interestingly, however, aneuploidy levels were significantly lower in the TP53-mut + group relative to the TP53-mut group. Therefore, aneuploidy levels may underlie the difference in IS expression between the two groups. To establish the association between TP53 mutation, co-mutation and aneuploidy levels in immune gene prediction set, we built multivariate regression models, adjusting the TP53 mutational and co-mutational status for the aneuploidy scores. In the multivariate models TP53 co-mutation (TP53-mut+) and aneuploidy were found to be independent predictors of the immune signature (Fig. 2E). A negative correlation was also found between aneuploidy and CTLA-4 and genes from PI3K\mTOR pathways (S-Fig. 8).
In line with the importance of the TP53 status potentially influencing response to ICI, an analysis of cell type composition, performed with Xcell software [14], revealed distinct immune cell composition across the three TP53 groups (Fig. 2F). In support of a fundamental difference between TP53-mut tumors with vs. without additional mutations, the abundance of 7 immune cell types was statistically different between TP53-mut and TP53mut + patients (S-Fig. 9).
STEP 2 - Phase II: Analysis of the immune gene sets and MYC dependent signature in two cohorts of HNSCC patients treated with PDL1-inhibitorsWe further investigated the role of the immune gene sets and of the TP53 mutated-dependent MYC dependent gene signature in two well characterized cohorts of HNSCC patients under treatment with PD-L1 inhibitors obtained from the GEO database (accession ID: GSE159067) and MSKCC dataset (cBioPortal.org [15],)
In Fig. 2G, we present the results of the analysis regarding the prognostic value of the immune gene sets. The left panel represents overall survival (OS), while the right panel represents progression-free survival (PFS). These results reinforce the findings from both the TCGA cohort and the CPTAC cohort (as shown in Fig. 1C, D, and E), providing further evidence that the expression-based immune signature (IS) score is associated with improved survival outcomes across multiple clinical datasets. The immune gene sets, we used to define our immune score, was also strongly correlated to the classification (“COLD” and “HOT”) introduced by Foy and colleagues (S-Fig. 10A). Notably, in Fig. 2H, left panel, low levels of the immune gene sets are significantly associated with stable or progressive disease during immunotherapy. Furthermore, low level of the TP53-dependent MYC signature was significantly associated with the immunologically “HOT” type (Fig. 2H, right panel). Furthermore, we conducted survival analysis on a cohort of 139 patients with Head and Neck cancer who underwent ICI treatment, and whose mutational status was established (MSKCC dataset). Despite the relatively modest sample size of the subgroups, it is apparent that even though the p-values may lack robustness, the disparity is striking. Specifically, the median survival among patients with co-mutations is twice that of patients solely harboring the TP53 mutation (10 months versus 5 months, respectively), as illustrated in the Fig. 2I. These results are in line with our finding on TCGA data and cell lines about the potential role of MYC in an immunogenic context. As further evidence supporting the role of co-mutational status, we assessed the expression of the 27-gene set used by Foy and colleagues to define their HOT score in TCGA HNSCC patients with different mutational statuses. Remarkably, we observed that co-mutated patients exhibited a “hotter” profile compared to TP53 mutated patients (S-Fig. 10B).
Finally, we used specific marker genes of the cell types identified in the cell enrichment analysis of TCGA data (Fig. 2F) to evaluate their quantitative expression on immunotherapy treated patients. Six out of the seven investigated cell types resulted strongly up-regulated in HNSCC patients characterized by complete or partial response to the treatment. The same cell types showed a significant reduced abundance in patients with low Immune Score (S-Fig. 10C).
Comments (0)