
記住我



For risk stratification and identification of genetic aberrations suitable as MRD targets, diagnostic material from 133 children with primary AML was examined (see “Methods” and Supplementary Table 3). The presence of selected fusion genes and mutations was prospectively investigated using (q)RT-PCR and sequencing. A primary genetic aberration was found in 102 children, while the remaining 31 children were further investigated using WTS. In 20 children, WTS identified rare or novel (presumably) primary genetic aberrations not included in the targeted screening, while seven children were found to possess fusion genes included in the targeted screening but not detected due to atypical variants.
The main genetic findings are summarized in Table 1 (for more details see Supplementary Table 3). A (presumably) primary genetic aberration was found in 97% of cases (129/133), and these AMLs are hereafter referred to as genetically classified. A large proportion of AMLs (81%) were classified into common subtypes: AML with KMT2A-r, PML::RARA, RUNX1::RUNX1T1, CBFB::MYH11, mutations (m) of GATA1, CEBPA or NPM1, respectively.
Table 1 Genetic and morphological (FAB) classification.Genetic aberrations that were identified as recurrent but rare in pediatric AML were found in 16 patients: UBTFm, RUNX1m, HOXA10 translocation, KAT6A::CREBBP, KAT6A::LEUTX, DEK::NUP214, BCR::ABL1, NUP98::NSD1 and CBFA2T3::GLIS2.
In five patients, fusion genes were identified, that have been described so far to occur sporadically (SPFQ::ZFP36L2 [18], XPO1::TNRC18 [19]) or not at all in AML (ETV6::CTNNB1, FUS::FEV, ZEB2::RUNX1). These genetic aberrations were assessed as (presumably) primary, based on the occurrence of these fusions (or fusions involving one of the partner genes) in hematological malignancies [20,21,22].
In the remaining four patients (3%), no primary genetic aberration was found, but only mutations that frequently occur as secondary in AML (FLT3-ITD, WT1m, KRASm, NF1m, PTPN11m and KITm; 1–3 mutations per patient) [23]. These four AMLs are hereafter referred to as genetically unclassified.
Applicability of gDNA-based MRD monitoringWe aimed to establish MRD monitoring at the gDNA level with a sensitivity of at least 10−4 (0.01%) in all patients. The preferred method for MRD monitoring was qPCR, while deep amplicon NGS was considered as a second option.
Three children from our cohort died shortly after AML diagnosis, MRD monitoring was thus relevant for 130 patients (“MRD cohort”), of which 126 had genetically classified AML. In 85 of them, the primary genetic aberrations were gene fusions. Targeted NGS (or PCR) was performed to identify genomic fusion sequences and succeeded in 82 of 84 cases examined. Of note, when analyzing WTS data (available in a proportion of patients), in approximately half of the cases the genomic fusion sequence could be found in retained introns of the fusion gene transcripts and targeted NGS was not necessary (data not shown). In all 82 cases with an identified genomic sequence, qPCR systems with the required sensitivity for target detection were implemented. In the two patients in whom genomic fusion was not found, fusion transcripts were used as MRD targets (NUP98::NSD1, ZEB2::RUNX1). One patient (SPFQ::ZFP36L2-positive) was not investigated for the fusion gene DNA sequence, because a TR gene rearrangement was used as a target for MRD monitoring (IG/TR rearrangements were specifically screened in this patients, based on the described occurrence of the SPFQ::ZFP36L2 fusion in a leukemia of T-cell origin [20]).
In 41/126 children with genetically classified AML, the aberrations available as targets for MRD monitoring were gene mutations (CEBPAm, NPM1m, GATA1m, UBTFm, RUNX1m) ranging from single base to complex ones. In 29 children, a quantification system with required sensitivity was implemented (in two of them NGS-based because of insufficient qPCR sensitivity). In four children (three with GATA1m and one with CEBPAm), sensitivity of detections was suboptimal (5 × 10−4) but still acceptable for MRD monitoring. In seven children, we were not able to detect primary aberrations (single base GATA1m) with sufficient sensitivity, and MRD was not monitored.
Similarly, in a single patient, we were not able to sensitively detect (presumably) primary RUNX1m, but the accompanying subclonal RUNX1m was used as MRD target with detection sensitivity of at least 10−4. This target was lost at AML relapse (Supplementary Fig. 3).
In 3/4 children with genetically unclassified AML, FLT3-ITD (n = 2) or WT1m (n = 1) were used as MRD targets (with detection sensitivity of at least 10−4), with the awareness of their potential subclonality.
In addition to the four children mentioned above, 1–2 secondary aberrations (WT1m, FLT3-ITD) were used as additional MRD targets in another six children with genetically classified AML and quantified in parallel with the primary aberrations (Supplementary Fig. 3). In four patients, the levels of all MRD targets correlated well, whereas in two patients the levels of secondary aberrations were consistent with a subclonal origin. These results illustrate the expected pitfalls of using secondary aberrations as targets for MRD monitoring.
Established quantification systems were used to monitor MRD in 122 children (representing 94% of the MRD cohort), in 120 children by qPCR and in two children by NGS. MRD was monitored using (presumably) primary aberrations as targets at the DNA level with a sensitivity of at least 10−4, or eventually 5 × 10−4, in 112 and four children, respectively (together representing 89% of the MRD cohort). In six children MRD was monitored at the DNA level, but using secondary aberrations as targets, or at the mRNA level with a sensitivity of at least 10−4.
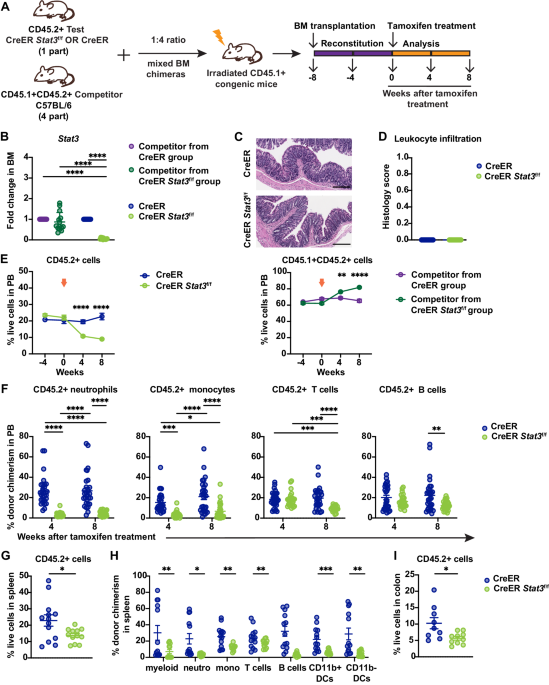
Diverse dynamics of MRD clearance in distinct AML subtypesThe vast majority of patients were treated according to the AML-BFM 2012 Registry protocol, where therapy consisted of 2 induction and 2–3 additional blocks of chemotherapy (CHT); BM for MRD detection was collected after each block (Supplementary Fig. 1). Therapy of children with DS-AMKL consisted of 4 blocks of CHT and the timing of BM sampling was similar; thus, these two groups of patients (106 patients with genetically classified AML in total) were analyzed together. Patients with APL were mostly treated according to different protocol, MRD clearance of APL is thus shown separately in Supplementary Fig. 4. A single patient with FLT3-ITD-positive NPM1m AML who was also not treated according to the AML-BFM 2012 Registry protocol was also excluded from the analyses described below.
Of all AML subtypes, patients with GATA1m AML had the fastest MRD clearance, 70% achieved molecular remission (mREM) after the first CHT block (at day 28; D28) (Fig. 1). Patients with prognostically favorable genetic subtypes (CBFB::MYH11, RUNX1::RUNX1T1, CEBPAm and NPM1m) were treated predominantly on the SR arm of the AML-BFM 2012 Registry protocol. Compared with GATA1m, their MRD clearance was significantly slower, none achieved mREM at D28 (70% vs. 0%, p < 0.0001), most patients (63–78% within individual subtypes) had MRD ≥ 10−3 at D28, and a significant proportion of patients (25–89% within individual subtypes) did not achieve mREM after the last CHT block. Patients with KMT2A-r AML and AML classified into remaining subtypes were treated predominantly on the intermediate- and high-risk (IR, HR) arms. In the subgroup with KMT2A-r, 33% (61%) achieved mREM at D28 (D56), thus their response to treatment was overall faster compared to the four prognostically favorable subtypes listed above (mREM 33% vs. 0% at D28, p < 0.0001). Importantly, except for CBFB::MYH11 AML, initial treatment (up to D56) on the SR, IR and HR arms was identical. There were no significant differences in MRD dynamics between patients with the two most common KMT2A-r (KMT2A::MLLT10 and KMT2A::MLLT3) while patients with other KMT2A-r had significantly slower MRD clearance (mREM at D28 44% in KMT2A::MLLT10/MLLT3 vs. 0% in other KMT2A-r, p = 0.02). Remaining patients, treated on the IR and HR arms, had various AML subtypes individually represented only in small numbers; when analyzed together as one genetically heterogeneous group, their response to treatment was worse compared to KMT2A-r AML, no patient achieved mREM at D28 (0% vs. 33%, p = 0.0018).
Fig. 1: Dynamics of MRD clearance.
The figure shows the dynamics of MRD clearance in patients with six different genetic subtypes of AML and patients with other AML subtypes (BCR::ABL1, CBF2A::GLIS2, DEK::NUP214, ETV6::CTNNB1, FUS::FEV, HOXA10-r, KAT6A-r, NUP98::NSD1, RUNX1m, SFPQ::ZFP36L2, UBTFm, XPO1::TNRC18, ZEB2::RUNX1) grouped together (OTHER). In AML with KMT2A-r, the dynamics of MRD clearance in three subgroups stratified by fusion partner genes is also shown. The Y-axis shows patient numbers, the X-axis shows treatment time points. #CHT was not administered either because it was not included in the respective treatment arm’s regimen or the patient relapsed/received modified therapy; *BM sampling was not performed or the time point was not reached.
Detection of molecular relapse by MRD monitoringIn order to detect early molecular relapse (mREL), MRD was monitored after treatment in a proportion of patients. To better understand the relevance of MRD measured in PB, together with some BM samples collected at different time-points during and after treatment, PB samples were also collected. The analysis confirmed published observations [24, 25] that MRD levels in PB may be (but not always are) lower compared to BM (Supplementary Fig. 5). Molecular relapse, defined as a reversal of negative MRD to positive or a 1-log increase in MRD positivity confirmed in subsequent sample, was observed in 11 patients (Fig. 2). In five patients, mREL as MRD 10−5 to 10−3 was detected in BM either during intensive treatment (n = 3) or after HSCT (n = 2). In six patients, mREL was detected in PB during post-treatment follow-up, in three of them as MRD ≥ 10−3. Hematological relapse followed in 13–86 days.
Fig. 2: Detection of molecular relapse.
The figure shows the course of MRD in 11 patients with detected molecular relapse. The unique numbers and primary aberrations of the patients are shown in the headers of the graphs. Primary aberrations were used as MRD targets in 10 patients, while the TCRD gene rearrangement was used as a target in patient UPN110. Three patients who relapsed while still on treatment are shown in the top row. Red circles correspond to BM samples, blue circles to PB samples. The Y-axis shows MRD levels, for graphical representation, non-quantifiable positive samples were assigned an MRD value of 1.00E−05. X-axis shows time since diagnosis (time 0) in days. gRisk, (cyto)genetic risk (see Supplementary Fig. 1 for risk stratification); Arm, treatment arm of AML-BFM 2012 registry protocol; *patients were treated in a different treatment arm than would correspond to gRISK based on clinicians’ decision; **gRISK was assigned retrospectively based on the corrected cytogenetic result reporting a complex karyotype, treatment followed the originally assigned IR gRISK; mREL molecular relapse, hREL hematological relapse, NEG negative, D day. None of the patients had WT1m; FLT3-ITD was present only in patient UPN080, who was enrolled in CPKC412A2218 trial and received FLT3-inhibitor.
Prognostic value of MRD monitoring at the gDNA levelWe investigated the prognostic value of gDNA-based MRD at early time-points of treatment. Given the excellent treatment outcomes of patients with APL, DS-AMKL, and patients treated on the SR arm of the AML-BFM 2012 registry protocol (n = 64, 5-years EFS 97%, median follow-up 3.2 years), we focused on patients treated on the IR and HR arms (n = 68, 5 years EFS 62%). This cohort involved nine patients who were reassigned from SR (n = 1) or IR (n = 8) to HR arm based on poor therapy response (see Supplementary Methods). Significantly different EFS and OS were observed between patients stratified by MRD levels 10−3 as well as 10−2 at D28 (Fig. 3). Patients stratified by MRD level 10−3 at D56 had significantly different OS only, while both EFS and OS were significantly different when patients were stratified by any positivity vs. negativity (Fig. 3). Neither (cyto)genetic risk nor treatment (IR vs. HR arm) had significant prognostic impact (Supplementary Fig. 6). In a multivariate analysis including (cyto)genetic risk and treatment, D28 MRD was the only significant predictor of outcome using both the 10−3 (p = 0.006 for EFS and 0.012 for OS) and 10−2 levels (p = 0.004 for EFS and 0.01 for OS) for stratification (Supplementary Table 4). In the same model but with D56 MRD, MRD was again the only significant predictor of outcome, whereas stratification by any positivity vs. negativity had stronger predictive value than stratification at 10−3 level (positivity vs. negativity: p = 0.004 for EFS and p = 0.022 for OS; 10−3 cut off: no significant difference for EFS, p = 0.029 for OS).
Fig. 3: Treatment outcomes of patients stratified by MRD levels.
The figure shows treatment outcomes for 64 patients treated in the IR and HR arms of the 2012 BFM AML Registry protocol stratified by MRD levels after 1st induction (D28) or 2nd induction (D56). A total of 68 patients were treated in the IR (n = 36) and HR (n = 32) arms, MRD was not measured in two of these patients, and the other two patients died before D28. Data on D56 MRD were missing in one patient (BM aspiration was not performed). Of the 64 patients included in the analyses, 35 patients had KMT2A-r AML, 26 patients had AML classified into one of the other 17 subtypes (1–3 patients per subtype), and 3 patients had unclassified AML. EFS event-free survival, OS overall survival, y year, NEG. negative, POS. positive. Censoring is indicated by crosses.
留言 (0)