
記住我

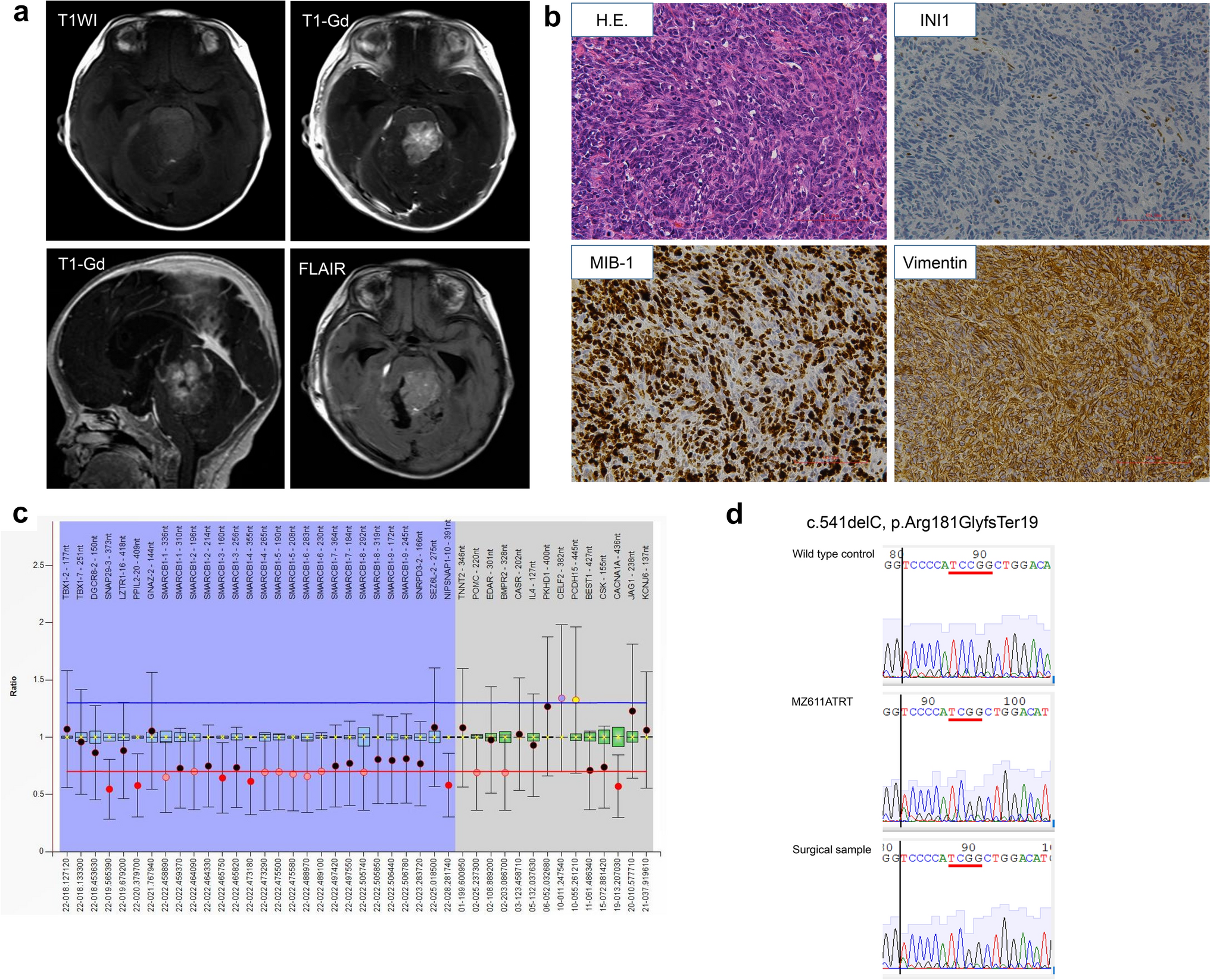
The donor patient was a 71-year-old male with hCCA. The patient had elevated tumor marker levels, and computed tomography (CT) images showed circumferential thickening and mild enhancement of the perihilar bile duct with luminal narrowing (Fig. 1a–c). A diagnosis of hCCA was considered. No preoperative radiotherapy or chemotherapy was administered, and the patient underwent resection of the hCCA. The tumor specimen showed a circumferential soft-tissue perihilar mass (Fig. 1d). The postoperative pathological diagnosis of this patient's tumor tissue was moderately differentiated adenocarcinoma of the bile duct (Fig. 1e).
Fig. 1
Clinical and pathological profile of CBC3T-1. Computed tomographic (CT) scan of the abdomen. Plain CT scan (a) showed circumferential thickening of the upper bile duct wall with luminal narrowing, while a contrast-enhanced CT scan (b, c) revealed that the tumor was slightly enhanced (yellow arrow). d General view of the surgically resected specimen. e H&E staining of primary tumor tissue. Scale bars 100 μm. f Clinicopathological profile of CCA patients
Cell cultureThe human eCCA cell line TFK-1 and human biliary epithelial cell line HIBEpiC were purchased from the National Biomedical Experimental Cell Resource Bank of China (Beijing, China). The cells were cultured in RPMI 1640 medium (Gibco) containing 10% FBS (Gibco, USA), 100-U/mL penicillin, and 100-mg/mL streptomycin (Gibco, USA), and placed in an incubator with a constant temperature of 37 °C and 5% CO2.
Primary cell cultureImmediately after surgery, specimens were obtained from the primary tumor tissue. The tissue was cut to a 1-mm3 size with scissors. Subsequently, the tissues were transferred to DMEM/F-12, (Gibco, USA) containing 200 U collagenase II and digested at 37 °C for 15–30 min, after which a single-cell suspension was obtained by filtering the supernatant through a 100-μm cell strainer. The filtrate was centrifuged (300 g/5 min), and the cell precipitate was resuspended in DMEM/F-12 supplemented with 10% FBS and 100-mg/ml Primocin™ (InvivoGen, CA). Cells were incubated at 37 °C in a humidified atmosphere with a 5% CO2 incubator. All subsequent experiments were performed after 30 generations.
Verification of cell lines by ATCCThe CBC3T-1 cell line was authenticated by ATCC. Briefly, genomic DNA was extracted from the CBC3T-1 cell line using a QIAamp DNA Mini Kit (Qiagen, The Netherlands). The alleles of 21 loci in CBC3T-1 cells (D19S433, D5S818, D21S11, D18S51, D6S1043, AMEL, D3S1358, D13S317, D7S820, D16S539, CSF1PO, Penta D, D2S441, vWA, D8S1179, TPOX, Penta E, TH01, D12S391, and D2S1338) were amplified by PCR and analyzed using an Applied Biosystems 3730xl DNA Analyzer (Applied Biosystems, USA). When the short tandem repeat (STR) data of CBC3T-1 cells in the databases of ATCC, DSMZ, and CELLOSAURUS were compared, the profile did not exactly match with any of the current data.
Cell Counting Kit (CCK)-8 cell growth assayCells were seeded into 96-well plates at a density of 6 × 103 cells per well. After incubation for 0, 24, 48, 72, and 96 h, Cell Counting Kit 8 (CCK-8) reagent (APExBio, USA) was added to the cells, and the cells were incubated for 2 h. The absorbance at 450 nm was measured.
Mycoplasma detection by PCRFor the detection of cellular mycoplasma, the medium of CBC3T-1 cells was collected and assayed according to the mycoplasma detection kit (Invitrogen, CA). DNA fragments were imaged under UV irradiation.
Chromosome karyotype analysisCells were incubated for 2 h using 0.4 µg/ml colchicine. Then cells were collected and incubated with 0.075 M KCl for 30 min (37 °C) and fixed 3 times with methanol: acetic acid (3:1) at room temperature for 10 min. Slides were then prepared and stained with Giemsa. Representative images of chromosome sets were obtained for karyotype analysis.
Live cell imagingCells were digested and cultured in 96-well plates until they attached to the surface. Live cell imaging was performed using a Cytation 1 imaging system (Biotek, USA) under a 4 × objective. Label-free live cell proliferation measurements were achieved by capturing two high-contrast brightfield images at each 2-h time point. Images were processed with Gen 5.
Spheroid formation assayCells were inoculated at a density of 1000 cells per well in an ultralow-attachment 96-well plate (Corning, USA) containing 10% FBS in DMEM/F-12. The cells were observed with a microscope on days 3, 7, and 14 of incubation.
Wound-healing assayCells were inoculated in 6-well plates. When 95% confluence was reached, the cell monolayers were scratched with a P-200 pipette tip, and the scratched monolayers were gently washed three times with phosphate buffer solution (PBS). Medium containing 10% FBS was then added for further incubation. Images were captured at 0, 12, and 24 h, and the distance between the two wound edges was quantitatively assessed by measuring the entire area of the scratches with ImageJ software.
Transwell migration/invasion assaysFor Transwell plate migration assays, a total of 8 × 104 cells were seeded in the upper chamber of an 8 μm Transwell plate (BD Biosciences, USA) with 100 μl of serum-free medium. In the lower chamber, 500 μl of medium containing 15% FBS was added. After 24 and 48 h of incubation, the cells in the upper chamber were carefully removed. Cells adhering to the membrane were fixed in 4% paraformaldehyde for 15 min and stained with 0.1% crystalline violet (Beyotime, China) for 15 min. For invasion assays, 50 µl of Matrigel (Corning, USA) diluted 1:4 with DMEM/F-12 was precoated in the upper chamber and seeded with 8 × 104 cells in 100 µl of DMEM/F-12. The rest of the procedure was similar to that of the Transwell migration assay.
Colony-forming assayCells were seeded in 6-well plates at a density of 700 cells/well in different complete media and allowed to form colonies for 14 days. Colonies were fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet for 15 min. The results were photographed and observed using an inverted phase contrast microscope as described above and analyzed using ImageJ software.
Sensitivity to anti‑cancer drugsFor testing of sensitivity to first-line clinical agents (oxaliplatin, cisplatin, gemcitabine, 5-fluorouracil, and paclitaxel) for CCA, CBC3T-1 cells were seeded at a density of 1 × 104 cells per well in 96-well plates. The cells were cultured overnight and then treated with anticancer drugs for 72 h. Cell viability was determined using a Cell Counting Kit 8 (APExBIO, USA).
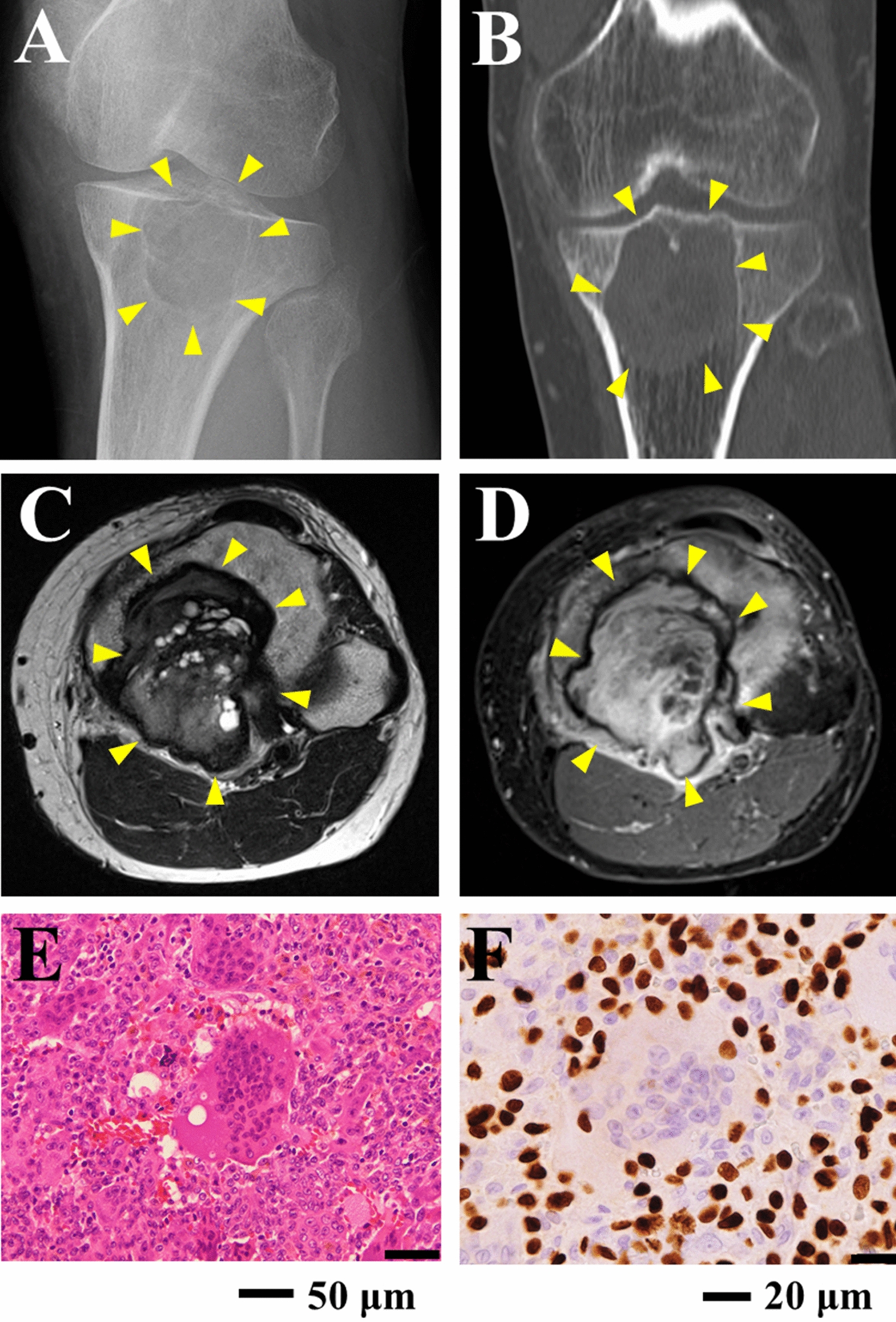
Tumorigenicity in NOD/SCID miceTo study the tumorigenicity of the CBC3T-1 cell line, 4–6-week-old female NOD/SCID (nonobese diabetic/severe combined immunodeficient) mice were purchased from Beijing Weitong Lihua Experimental Animal Technology Co. Ltd. A total of 2 × 106 cells were resuspended in 0.2 ml of DMEM/F-12 and Matrigel (Corning, USA) mixture and injected subcutaneously into each NOD/SCID mice. The animals were kept in a laminar flow cabinet under specific pathogen-free conditions. The mice were continuously monitored for tumor growth. After 21 days, tumor tissue was collected, measured and weighed, fixed in 10% formalin, embedded in paraffin, stained with hematoxylin and eosin (H&E), and subjected to immunohistochemistry (IHC). The animal experiment was performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of China. The protocol was approved by the Ethics Committee of the First Hospital of Lanzhou University (Approval No. LDYYLL-2022-506).
H&E staining and IHCThe samples were embedded in paraffin and cut into 4-μm-thick sections. The sections were dewaxed in xylene, hydrated in a graded alcohol series, and washed with phosphate-buffered saline for H&E staining. For IHC analysis, the slides were heated with 10 mM sodium citrate (pH 6.5) in a pressure cooker for 15 min. The nonspecific antigens were blocked with catalase enzyme body for 10 min, and 10% normal goat serum was added for 10 min. Then, the slices were incubated overnight at 4 °C with primary antibodies against CK7 (MAB-0828, 1:300, China), CK19 (MAB-0829, 1:300, China), Ki67 (MAB-0672, 1:1000, China), and p53 (MAB-0674, 1:500, China). Excess primary antibodies were washed off with PBS, and then, the slices were incubated with secondary antibodies for 30 min for DAB color development. Finally, counterstaining was performed with hematoxylin. The slides were observed using an Olympus DP26 light microscope.
RNA sequencing analysisTo explore the transcriptome changes in CBC3T-1 cells, normal bile duct cells HIBEpiC were used as a control. Total RNA was extracted from frozen cell pellets using an RNeasy Micro Kit (Qiagen, CA) according to the manufacturer’s protocol. Then, RNA sequencing (RNA‐seq) was performed using the BGISEQ‐500 platform at BGI Genomics (Wuhan, China). The library preparation followed BGI’s standard procedure.
Differential gene expression analysisDifferentially expressed genes (DEGs) were filtered and analyzed according to the following criteria: condition setting (|fold change|> = 2, Q value < 0.05). We used the BGI online platform (https://biosys.bgi.com/) to analyze the Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways of the DEGs.
Whole‑exome sequencing (WES) of CBC3T-1 cellsSequencing and data analysis were performed at BGI (Wuhan, China). Briefly, CBC3T-1 cells were aligned with samples of adjacent normal tissue from this patient’s resected tumor tissue and genomic DNA extraction was performed. Library construction and whole‑exome capture of genomic DNA were performed using SureSelect Human All Exon V6 (Agilent), and the captured DNA library was sequenced on the DNBSEQ platform. Clean reads were aligned to the reference human genome (build hg19) using the Burrows‒Wheeler Aligner. To identify somatic mutations, we compared CBC3T-1 cells with adjacent normal tissue, filtering out germline mutations in normal tissue and retaining only those somatic mutations found in tumor cells during the analysis.
Driver gene analysisWe compared somatic mutations with known driver genes in databases and the literature and screened out known driver genes in tumor samples. The reference data sources were Integrative OncoGenomics (IntOGen), Cancer Gene Census (CGC), three highly cited articles [14,15,16], and pan-cancer data [17].
Statistical analysisStatistical significance was calculated by unpaired two-tailed Student’s t test using GraphPad Prism 8 (GraphPad Software, Inc.). The data are presented as the means and standard deviations (SD) from at least three replicate analyses (bars). P < 0.05 was considered to indicate a statistically significant difference.
留言 (0)