
Remember me





Enrollment began in February 2020 but was slower than expected because of the COVID-19 pandemic. To minimize delay in the clinical development of amcenestrant, an unplanned administrative interim analysis was performed after 63 study participants had been randomized. This analysis, which included 55 evaluable participants, provided pharmacodynamic and safety data that were deemed sufficient to enable the strategic development decisions for which AMEERA-4 had been designed and did not change the benefit-risk profile of amcenestrant. Consequently, the decision was taken to terminate the trial before it was fully enrolled.
As a result of early termination of the trial, no formal statistical inferences were conducted; only descriptive statistics were provided.
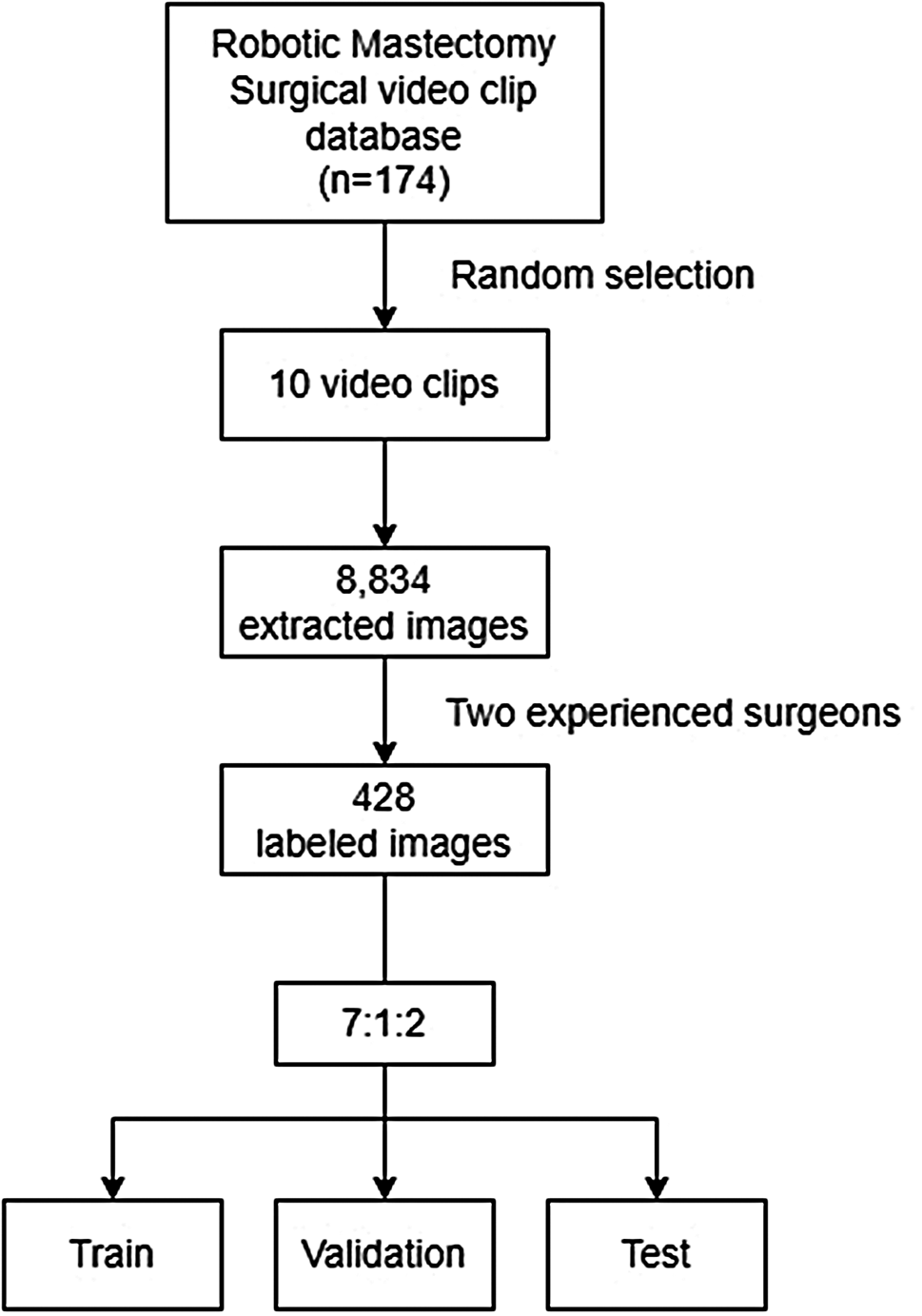
Participant dispositionWhen enrollment was stopped in April 2021, 135 participants had been screened, and 105 had been randomized: 34 to amcenestrant 400 mg, 36 to amcenestrant 200 mg, and 35 to letrozole (Fig. 1). All participants in the amcenestrant 200 mg and letrozole arms of the study completed the treatment period. One participant randomized to receive amcenestrant 400 mg withdrew consent before receiving their first dose of study medication. Thus, there were 104 participants in the safety population. The mITT population included 95 of the 105 randomized participants. The 10 participants who were excluded from the mITT population comprised 9 participants who had incomplete pre- and/or post-treatment Ki67 data (amcenestrant 400 mg: n = 2; amcenestrant 200 mg: n = 1; letrozole: n = 6), and the participant described previously who withdrew consent before receiving any study medication.
Fig. 1
CONSORT diagram. Abbreviations: ITT, intent-to-treat; mITT, modified intent-to-treat
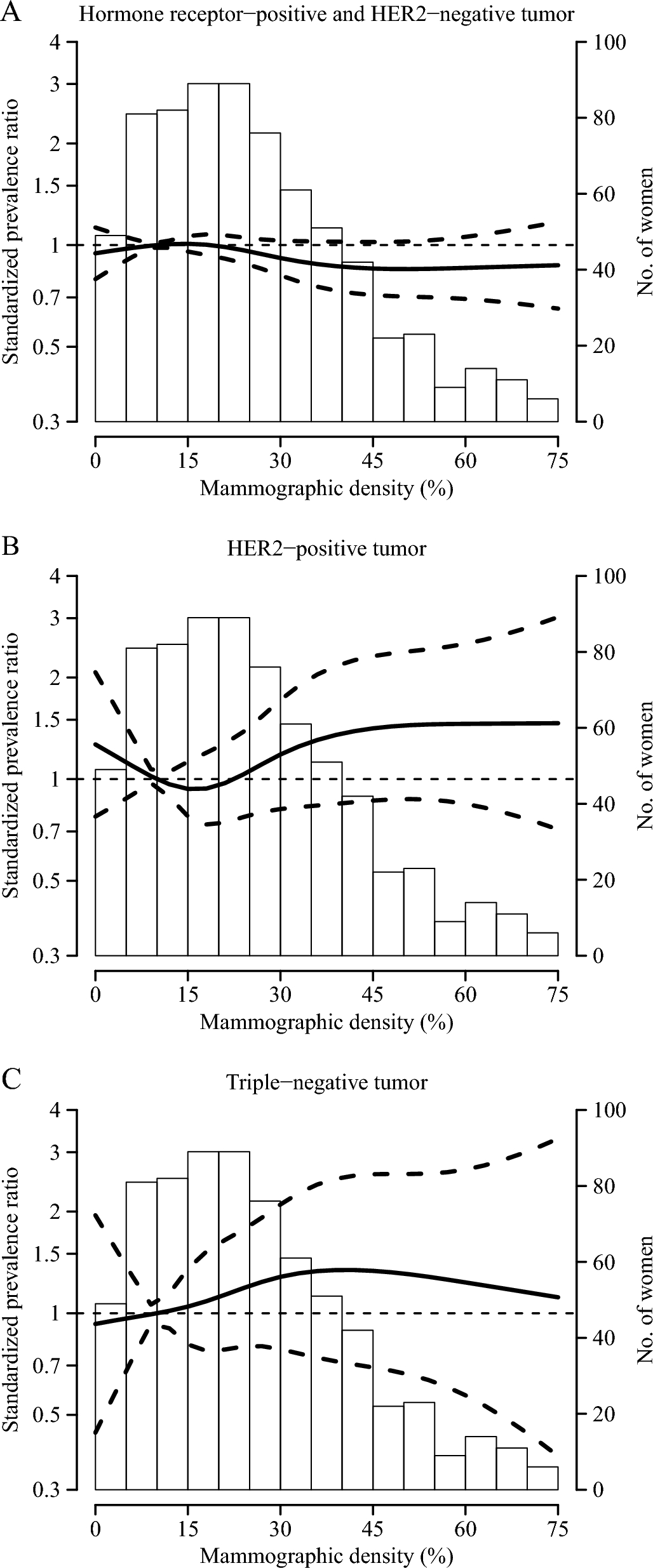
Participant and disease characteristicsThe characteristics of the study population were as expected, and there were no major imbalances between the treatment arms (Table 1). In the study population as a whole, median age and body weight were 62.0 years and 71.7 kg, respectively. Most participants were white, and all but three (one in each study arm) had stage I or II breast cancer. In accordance with the inclusion criteria, all participants had luminal B disease (defined as ER+, PgR±, and Ki67 ≥ 15%) by local assessment; 94 (89.5%) had PgR+ tumors.
Table 1 Baseline demographics and disease characteristics per local assessmentThe representativeness of the study population can be evaluated with reference to the information summarized in Additional file 1: Table S2.
Disease characteristics per central assessment are shown in Additional file 1: Table S3. Eleven percent (11/100) of participants across treatment groups had Ki67 < 15% (i.e., were classed as having luminal A disease), with less prevalence in the amcenestrant 200 mg group (2/36 participants; 5.6%) than in the amcenestrant 400 mg group (4/32 participants; 12.5%) or letrozole group (5/32 participants; 15.6%). The proportion of participants with Ki67 ≥ 20% was higher in the amcenestrant 400 mg group (26/32 participants; 81.3%) than in the amcenestrant 200 mg group (26/36 participants; 72.2%) or letrozole group (22/32 participants; 68.8%).
Median ER and PgR H-scores at baseline, per central review, were 300.0 and 120.0, respectively, in the amcenestrant 400 mg arm; 295.0 and 110.0, respectively, in the amcenestrant 200 mg arm; and 299.0 and 172.5, respectively, in the letrozole arm (Additional file 1: Table S3).
OutcomesIn the safety population (n = 104), the median relative dose intensity (defined as [actual dose intensity/planned dose intensity] × 100, where planned dose intensity [mg/day] = planned dose at Day 1) was 100% in all three treatment arms. Only one episode of dose modification was reported; this was due to participant error and occurred in the 200 mg arm. Three participants (one in the 200 mg arm and two in the 400 mg arm) had a total of four episodes of dose omission (one episode due to COVID-19, two because the participant forgot, and one unexplained). All participants underwent surgery for breast cancer as planned, between one and three days after the last dose of study medication. Ninety-two participants (88.5%) underwent surgery the day after their final dose.
Pharmacodynamics and pharmacokineticsDecreases in Ki67 expression were observed in most participants in all treatment groups (Fig. 2). Geometric mean Ki67 values at baseline and Day 15, and relative change from baseline, are summarized in Table 2. The geometric LSM estimates for relative change from baseline in Ki67 were − 75.9% (95% CI − 81.9 to − 67.9) for amcenestrant 400 mg, − 68.2% (95% CI − 75.7 to − 58.4) for amcenestrant 200 mg, and − 77.7% (95% CI − 83.4 to − 70.0) for letrozole. The geometric LSM ratio of proportional change versus letrozole was 1.08 (95% CI 0.72–1.63) for amcenestrant 400 mg and 1.42 (95% CI 0.95–2.12) for amcenestrant 200 mg (ratios < 1 favor amcenestrant). The proportion of participants with a ≥ 50% reduction in Ki67 versus baseline was 74.2% (95% CI 55.4–88.1) in the amcenestrant 400 mg arm, 68.6% (95% CI 50.7–83.1) in the amcenestrant 200 mg arm, and 89.7% (95% CI 72.6–97.8) in the letrozole arm. Twelve participants (5 in the amcenestrant 400 mg arm, 3 in the amcenestrant 200 mg arm, and 4 in the letrozole arm) had complete cell cycle arrest (CCCA), defined as post-treatment Ki67 ≤ 2.7%.
Fig. 2
Absolute change in Ki67 from baseline to Day 15 per central review, by baseline Ki67 (mITT population). Each colored line sloping from left to right represents an individual study participant. Reading from top to bottom, the box-and-whisker plots indicate: the highest observation within the range of Q3 and Q3 + 1.5 × (Q3–Q1); Q3; the median value; Q1; and the lowest observation within the range of Q1 and Q1–1.5 × (Q3–Q1). Abbreviations: mITT, modified intent-to-treat; Q, quartile
Table 2 Pharmacodynamic results (mITT population, n = 95)Individual changes in ER H-score are shown in Fig. 3. The LSM estimate (95% CI) of the absolute change in ER H-score from baseline, based on central assessment, was − 176.7 (− 201.4 to − 152.0) in the amcenestrant 400 mg arm and − 202.9 (− 226.1 to − 179.7) in the amcenestrant 200 mg arm (Table 2). One participant in the amcenestrant 400 mg arm had a high percentage change in ER H-score (+ 215.8%) that impacted the overall result for the group. The median relative reduction in the ER H-score was 65.3% in the amcenestrant 400 mg arm and 68.3% in the amcenestrant 200 mg arm. In contrast, and as predicted by its mechanism of action, letrozole was associated with modest reductions in ER H-score from baseline to Day 15 (mean absolute change, − 32.5; median relative change, − 9.5%).
Fig. 3
Absolute change in ER H-score from baseline to Day 15 per central review (mITT population). Each colored line sloping from left to right represents an individual study participant. Reading from top to bottom, the box-and-whisker plots indicate: the highest observation within the range of Q3 and Q3 + 1.5 × (Q3–Q1); Q3; the median value; Q1; and the lowest observation within the range of Q1 and Q1–1.5 × (Q3–Q1). Abbreviations: ER, estrogen receptor; mITT, modified intent-to-treat; Q, quartile
PgR H-scores also decreased from baseline to Day 15 in all treatment groups (Table 2 and Additional file 1: Figure S2). The LSM (95% CI) of absolute change from baseline was − 58.2 (− 78.4 to − 38.1) in the amcenestrant 400 mg arm, − 68.3 (− 87.4 to − 49.2) in the amcenestrant 200 mg arm, and − 88.4 (− 110.1 to − 66.6) in the letrozole arm. The median relative changes from baseline were − 70.0%, − 74.4% and − 75.3%, respectively.
Amcenestrant plasma concentrations were measured in 30 participants in the amcenestrant 400 mg arm and 27 participants in the amcenestrant 200 mg arm. On Day 14, geometric mean plasma concentrations were 258 ng/mL at pre-dose, rising to 2228 ng/mL at 3 h post-dose in the 200 mg group, and 452 ng/mL at predose, rising to 3399 ng/mL in the 400 mg group. These concentrations showed an increase of systemic exposure between the 200 mg and 400 mg doses (Additional file 1: Table S4).
SafetySixteen participants (48.5%) in the amcenestrant 400 mg arm, 16 participants (44.4%) in the amcenestrant 200 mg arm, and 18 participants (51.4%) in the letrozole arm experienced at least one TEAE of any grade during the treatment and post-treatment periods (Table 3). Almost all TEAEs were of Grade 1 or 2 severity. Two participants, both of whom were in the amcenestrant 200 mg arm, experienced a TEAE of Grade 3 or higher (one case of pneumonia and one of wound infection). These AEs required hospitalization and were therefore considered serious but were not considered to be treatment related. No participants discontinued treatment due to a TEAE, and there were no deaths either during the treatment or follow-up periods.
Table 3 TEAEs and TRAEs reported in ≥ 5% of study participants in one or more study armsSeven participants (21.2%) in the amcenestrant 400 mg arm, 8 participants (22.2%) in the amcenestrant 200 mg arm, and 9 participants (25.7%) in the letrozole arm experienced at least one TRAE of any grade during the treatment and follow-up periods (Table 3). There were no serious TRAEs in any arm, nor any TRAEs of Grade ≥ 3 severity. The most commonly reported TRAEs (i.e., reported in ≥ 5% of participants in at least one study arm) were hot flush, headache, feeling cold, arthralgia, asthenia, and diarrhea. Hot flush, arthralgia, and diarrhea were reported by more participants in the letrozole arm than in either of the amcenestrant arms. TRAEs reported in a single participant in ≥ 1 treatment arm were abdominal distension, alopecia, constipation, decreased appetite, dry skin, dyspepsia, fatigue, increased ALT, insomnia, lower abdominal pain, musculoskeletal pain, musculoskeletal stiffness, myalgia, nausea, night sweats, pain in extremity, pollakiuria, and upper abdominal pain.
Three participants each had a single pre-specified AESI, all of which were non-serious Grade 2 increases in ALT. All three participants had received amcenestrant: one had received 400 mg, and the other two had received 200 mg. However, only the case at 400 mg was considered to be related to study drug. All cases resolved without sequelae and did not necessitate treatment interruption or dose modification. No bradycardia (pulse rate < 50 beats/min) or eye disorders, other than a single case of dry eye that was unrelated to study treatment, were reported in any study group.
Genomics and transcriptomicsCell cycle score signatures decreased in all three treatment groups (Additional file 1: Table S5 and Figure S3). The median change from baseline was − 0.83 in the amcenestrant 400 mg arm, − 0.56 in the amcenestrant 200 mg arm, and − 0.84 in the letrozole arm. Changes in molecular subtype from baseline to Day 15, assessed using the Prosigna® gene expression assay (PAM50), are summarized in Additional file 1: Figure S4. In all three groups, randomized treatment was associated with an increase in the number of participants with luminal A disease and a simultaneous decrease in the number with luminal B disease.
Mutational profiling of tumor DNA was performed at baseline in 75 participants (26 participants in the amcenestrant 400 mg arm, 28 participants in the amcenestrant 200 mg arm, and 21 participants in the letrozole arm [Additional file 1: Figure S5]). The most commonly mutated genes at baseline were PIK3CA (36.0%), TP53 (22.7%), and GATA3 (17.3%). Mutant BRCA1 and BRCA2 were each identified in 4 participants (5%), including one participant who had mutations in both genes. Data on baseline mutations in cfDNA were available for 92 participants, 26 of whom had wild-type DNA and 66 of whom had mutations (24 participants in the amcenestrant 400 mg arm, 23 participants in the amcenestrant 200 mg arm, and 19 participants in the letrozole arm [Additional file 1: Figure S6]). The most common mutations were in the TP53 and GNAS genes (22.7% and 9.1% of participants, respectively).
Comments (0)