
記住我
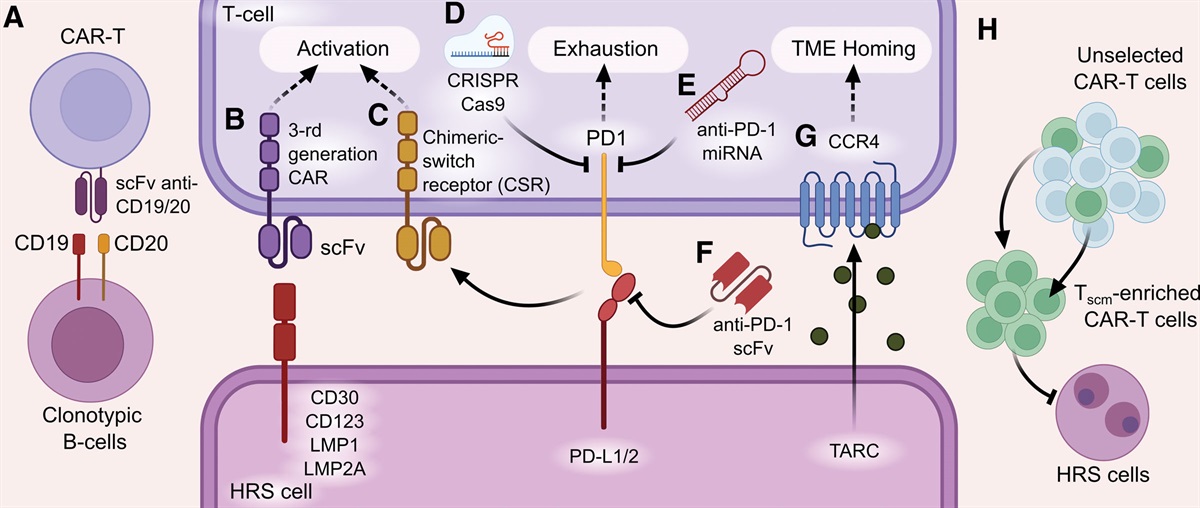


With aging, acquired genetic alterations accumulate in hematopoietic stem or progenitor cells. Positive selection may provide a growth advantage for clones carrying driver mutations, and subsequent clonal expansion is called clonal hematopoiesis.1 Mosaic loss of chromosome Y (mLOY) is the most commonly acquired cytogenetic alteration in aging male individuals from the general population.2,3 mLOY associates with all-cause mortality and cardiovascular disease (CVD).3,4 Driver gene mutations (GMs) are observed in 10%–60% of community-dwelling individuals ≥60 years and predispose to hematological malignancies and CVD.1,5–7 Although both mLOY and GMs occur at higher age and share implications for otherwise healthy individuals, their relationship has not been systematically investigated. Previously, in a study comprising 24 community-based individuals, a significant enrichment of GM has been observed in males with mLOY and a higher cell fraction of mLOY was found in males carrying multiple GMs.8 Moreover, a co-occurrence between mLOY with cellular fractions ≥75% and GM was reported in 73 patients presenting in the clinic for cytogenetic and molecular evaluation.9 We aimed to investigate the association between mLOY and GM in a large population-based cohort and their combined implications for the risk of clonal progression and all-cause mortality.
This study was performed within the population-based Lifelines Cohort, a study examining 167,729 persons living in the North of the Netherlands.10,11 The University Medical Center Groningen Medical Ethical Committee approved the Lifelines protocol under 2007/152, and the study was conducted in accordance with the Declaration of Helsinki.
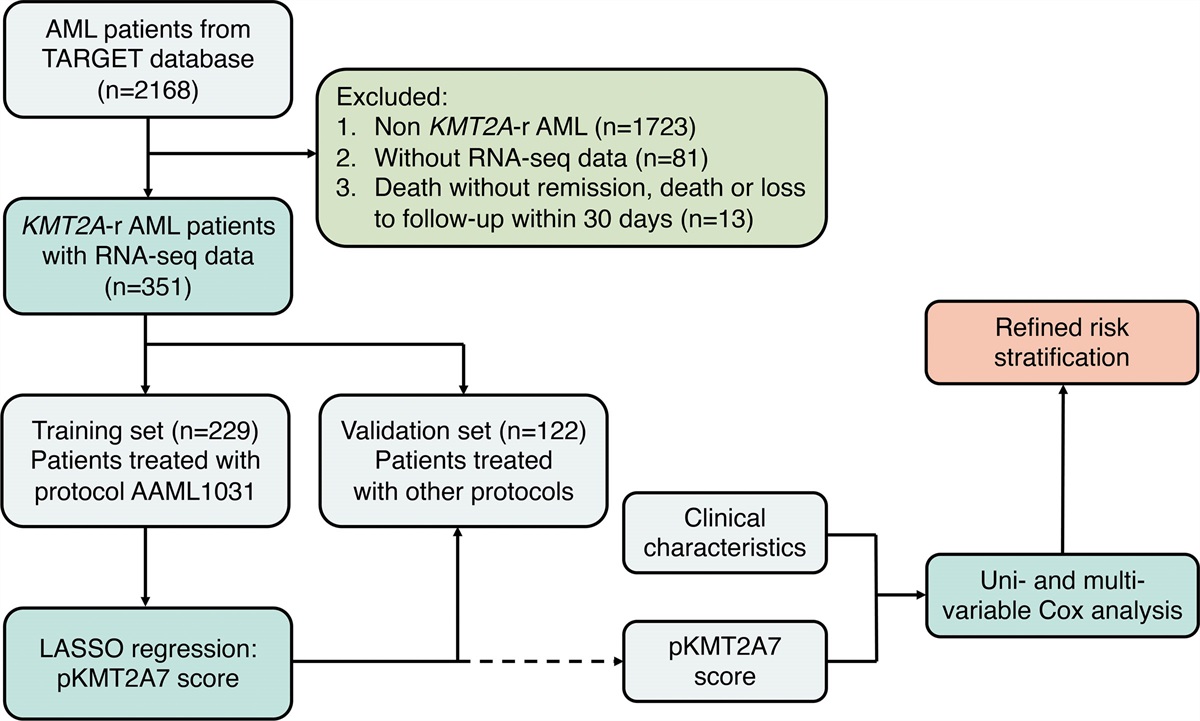
We included (1) a subcohort of 15,098 males with available single-nucleotide polymorphism array genotype data to detect mLOY and (2) a subcohort of 2463 male individuals aged ≥60 years for whom targeted error-corrected next-generation sequencing (NGS) for 27 genes to detect GM was previously performed (Figure 1A). The latter cohort was enriched for blood count abnormalities (Suppl. Figure S1).12–14
 Figure 1.:
Figure 1.: Cohort overview and coexistence of mLOY and GM. (A) Flowchart of cohort selection within the complete Lifelines cohort (n = 167,729). (B) Prevalence of mLOY per age group for all males with genotype data (n = 15,098). (C) Prevalence of all GM (dark blue), DNMT3A mutations (red), TET2 mutations (light blue), and ASXL1 mutations (green) per age group for all males with NGS data (n = 2463). (D) Distribution of highest VAF per individual and cellular fraction for all detected GM and mLOY events, respectively. (E) Venn diagram displaying the overlap between males with mLOY (green) and GM (blue) in males with genotype and NGS data (n = 538). (F) Mutational landscape for males according to the presence of mLOY (indicated in green). Each column represents 1 individual and multiple driver mutations in the respective genes are represented in dark blue. (G) Diagram for co-occurrence and mutual exclusivity of mLOY and recurrent driver GMs. Odds ratios and P-values derived from Fisher’s exact test are displayed. (H) Distribution for the maximum affected cell fraction in males with mLOY (green), GM (blue) and males carrying both lesions (turquoise). For GM, assuming heterozygosity, the cellular fraction was calculated by duplicating the variant allele frequency. (I) Distribution of age in males with mLOY (green), GM (blue), and males carrying both lesions (turquoise). (J) Violin plot displaying the number of mutated genes in males with and without mLOY. (K) Cellular fraction of the mLOY clone for males with 0, 1, and ≥2 GM. (L) Scatter plot showing the cellular fraction for combinations of mLOY and GM detected in the same individual. Dots above the dashed line fulfill the pigeonhole principle. One individual can appear multiple times in this plot when multiple GM are present. Spliceosome GMs include SF3B1, SRSF2, and U2AF1. cf = cellular fraction; CH = clonal hematopoiesis; GM = driver gene mutations; mLOY = mosaic loss of chromosome Y; NGS = next-generation sequencing; OR = odds ratio.
For the detection of mLOY, we used the Mosaic Chromosomal Alterations (MoChA) software (https://github.com/freeseek/mocha).15,16 In short, the software uses haplotype phasing information to detect chromosomal mosaic aberrations that create an allelic imbalance in the inherited 1:1 ratio of maternal and paternal chromosomal segments. A hidden Markov-model-based approach is applied to detect allelic imbalances at heterozygous sites, including pseudo-autosomal regions (PARs) of the sex chromosomes. The detection limit of MoChA for mLOY approximates 1% cellular fraction. Genotyping was performed using Infinium Global Screening Array Multiethnic Disease Version 1.0 for a subcohort of Lifelines participants (n = 36,339 including n=15,098 males that passed central quality check procedures). This array covers 576 markers in the PAR1/2 regions. Raw probe intensity data were used for genotype calling and normalization using the proprietary Illumina GenCall algorithm. The gtc2vcf plugin for bcftools (https://github.com/freeseek/gtc2vcf) was used to convert the resulting genotype files to VCF format. Next, variants that fell within segmental duplications with low divergence (<2%), variants with a high level of missingness (>3%), with a discordance between genotype and sex, or with excess heterozygosity were removed. Long-range statistical haplotype phasing was performed using SHAPEIT4 and phased genotypes were ligated using bcftools concat (https://github.com/samtools/bcftools). We used pedigree information to improve phasing accuracy with the plugin trio-phase for bcftools (https://github.com/freeseek/mocha). After applying MoChA to phased genotypes, samples with phased B allele frequency autocorrelation >0.03 (indicative of contamination or of other potential sources of poor DNA quality) were excluded and we restricted to mLOY calls with base pair length of ≥2 Mb and relative coverage estimate <2.5.
Targeted error-corrected NGS to detect GM was performed for a subcohort of 4715 Lifelines participants ≥60 years (including n = 2463 males) using a panel of 27 myeloid- and lymphoid-associated driver genes (Suppl. Table S1), as described before.17 We applied a variant call threshold of 1% variant allele frequency (VAF) and with at least 10 consensus reads.
All statistical analyses were performed using R version 4.0.2. Testing for associations between GM or specific driver GMs and mLOY was performed using Fisher’s exact test, with additional correction for age and blood count abnormalities in multivariable logistic regression analyses. Two-group nonparametric data were statistically compared using the Mann-Whitney U test. The Jonckheere-Terpstra test was used to assess differences in the cellular fraction of mLOY for an increasing number of GM. The Spearman correlation coefficient was used to describe linear relationships between nonparametric variables. Multivariable linear regression was used to compare the clone size between subgroups with age as covariable. We retrieved data on development of hematological malignancies by linkage to the Netherlands Cancer Registry (IKNL), with nationwide coverage of ≥95% of all newly diagnosed malignancies confirmed by histology and/or cytology. Individuals with a recorded previous diagnosis were excluded from analysis. Cumulative incidence graphs were constructed using the Aalen-Johansen estimator, with death as a competing risk. Cox regression was used to calculate hazard ratios (HRs) and 95% confidence intervals (CIs) for risk of death, corrected for age. All statistical tests were performed 2-sided, and P-values <0.05 were considered significant.
We detected mLOY in n = 1104 out of 15,098 genotyped males. The increasing prevalence of mLOY with age (42.6% in males ≥70 years; Figure 1B) corresponded to a previous study using comparable methodology.2 Among 2463 males with NGS data, the frequency of GM, including most commonly mutated genes DNMT3A, TET2, and ASXL1, increased with aging (Figure 1C). We thus confirm that both mLOY and GM emerge with advancing age in the general population.2,5 mLOY was detected at a median cellular fraction of 5.8% (min-max, 2.7%–67.7%; Figure 1D). For GM, median highest VAF per individual was 3.1% (min-max, 1.0%–86.0%; Figure 1D). For 538 male individuals aged ≥60 years, data on both mLOY and GM were available. mLOY and GM were detected in 178 and 205 males, respectively, with both mLOY and GM being present in n = 74 (24%). There was no significant co-occurrence of mLOY and GM (P = 0.258; Figure 1E). The mutational landscape was comparable for cases with and without mLOY, and no significant co-occurrence between the most commonly mutated genes and mLOY was observed (DNMT3A OR, 0.84 [95% CI, 0.54-1.29], P = 0.467; TET2 OR, 1.26 [95% CI, 0.71-2.19], P = 0.412; ASXL1 OR, 1.27 [95% CI, 0.32-4.48], P = 0.767; JAK2 OR, 1.01 [95% CI, 0.09-7.13], P = 1.000; Spliceosome OR, 1.76 [95% CI, 0.48-6.21], P = 0.373; Figure 1F, G). Additional correction for age and presence of blood count abnormalities showed similar results (Suppl. Table S2A). Thus, mLOY and GM may emerge independently with aging. We speculate that different selective pressures may underlie the acquisition of genetic aberrations or expansion of clones with mLOY and GM.
While others have found significant co-occurrence of mLOY and GM,7–9 we did not find such a relationship in our cohort. Differences between the studies included cohort sizes (varying between n = 24 and n = 6127) and different methods used to call mLOY (bioinformatic pipelines and conventional karyotyping). Our study was limited by a relatively small sample size and the fact that our dataset mostly included individuals with small mLOY clones. We performed subgroup analyses for the association between mLOY and GM clones with higher cellular fractions (Suppl. Table S2B). Interestingly, individuals with mLOY at higher cellular fractions (≥5%) were at higher risk to carry GM (using VAF cutoff ≥1%). Copresence might only be evident for mLOY clones with substantial expansion (eg, larger clonal size).9 We may speculate that NGS screening seems not indicated for males with established small mLOY clones in the absence of other indications. It should be noted that, using extremely sensitive assays, the frequency of detection for mLOY and GM increases and the copresence of even smaller clonal events needs to be determined. Finally, the co-occurrence of mLOY and GM and its clinical significance in specific or high-risk groups, including those with peripheral blood count abnormalities and the oldest old, needs to be established.
Next, we questioned whether the copresence of mLOY and GM in aging males correlates with enhanced clonal expansion. Interestingly, a significantly higher clonal size was detected in males carrying both lesions as compared with those carrying only mLOY or GM (median cellular fraction 9.1% versus 6.4% for GM and 6.6% for mLOY; P < 0.001), also after correction for age (GM versus both P = 0.033; mLOY versus both P = 0.002; Figure 1H, I). The number of mutated genes was not different for males with mLOY versus those without (P = 0.409; Figure 1J). For individuals with multiple GM, we observed a higher clone size for mLOY (P = 0.038; Figure 1K), which corresponds to previous work (n = 73).9 These results suggest a combinatorial effect on clonal expansion for the small group of individuals with co-occurrent mLOY and GM. There was, however, no correlation between the clone size of detected GM and mLOY (Spearman’s correlation coefficient −0.037; P = 0.753). Although single-cell sequencing is the method to prove that genetic events occur in the same cell, a sum of mutated cell fractions exceeding 100% is indicative for presence of genetic events in the same cell population (pigeonhole principle).18 None of the events in our cohort complied with this principle (Figure 1L), mainly explained by the low cellular fractions of most events.
A higher risk of incident hematological malignancies was previously reported for mLOY and GM.1,9 In line with this, we observed higher cumulative incidences of hematological malignancies for males with mLOY (P < 0.001) and GM (P = 0.004). GM were associated with a higher incidence of myeloid malignancies, while mLOY associated with incident lymphoid malignancies (Figure 2A–D). This may partly be explained by our gene panel, which mainly included myeloid malignancy-associated driver genes. Due to a small number of events in the overlap cohort (n = 2), it was not possible to study the risk for hematological malignancies in individuals carrying mLOY and GM.
 Figure 2.:
Figure 2.: Malignant transformation and prediction of all-cause mortality by mLOY, GM, and both lesions. (A) Cumulative incidence of myeloid malignancies for males with and without mLOY. (B) Cumulative incidence of lymphoid malignancies for males with and without mLOY. (C) Cumulative incidence of myeloid malignancies for males with and without GM. (D) Cumulative incidence of lymphoid malignancies for males with and without GM. (E) Forest plot to show the risk of all-cause mortality with 95% CI for males with mLOY (green), GM (blue), and males carrying both lesions (turquoise). Absence of the respective abnormality was used as a reference. P-values for cumulative incidence of hematological malignancies were reported from Gray’s test. CI = confidence interval; GM = driver gene mutations; mLOY = mosaic loss of chromosome Y.
Both mLOY and GM have been reported to associate with all-cause mortality,1,4,5 but the combinatorial effect of these lesions on survival is unknown. In our population-based cohort, GM was associated with inferior OS (HR, 1.36; 95% CI, 1.08-1.71; P = 0.009; Figure 2E). In contrast to previous work,4 the presence of mLOY did not affect the risk of death in our cohort (P = 0.934; Figure 2E). Differences in cohort characteristics (eg, our study was restricted to males aged ≥60 years with potential survivorship bias) and differences in detection method for mLOY may explain this contradiction. There was no significant interaction effect of mLOY and GM on the risk of death (P = 0.853).
In conclusion, we did not observe a co-occurrence of mLOY and GM in a population-based cohort of older males. The majority of small-sized clonal lesions presumably emerge independently with aging. However, co-occurrence of mLOY and GM is characterized by higher mutant cell fractions. Future studies may investigate the copresence of mLOY and GM in mutated cells and their joint role in malignant progression.
ACKNOWLEDGMENTSThe authors wish to acknowledge the services of the Lifelines Cohort Study, the contributing research centers delivering data to Lifelines, and all the study participants. They thank Gerben van der Vries and Marloes Benjamins-Stok for their help concerning the raw UGLI data. They further thank Esteban Lopera and Serena Sanna for performing the initial UGLI sample QC. Further, the authors would like to thank the University Medical Center Groningen (UMCG) Genomics Coordination Center, the UG Center for Information Technology, and their sponsors BBMRI-NL and TarGet for storage and computing infrastructure. The authors thank the registration team of the Netherlands Comprehensive Cancer Organization (IKNL) for the collection of data for the Netherlands Cancer Registry (NCR) as well as IKNL staff for scientific advice.
AUTHOR CONTRIBUTIONSPK, IAvZ, AOdG, and JBS contributed to data collection, analysis, and interpretation of the data. IAvZ and AOdG contributed to study design. GG designed MoChA software. AGD, BAvdR, and JJS were involved in the interpretation of the data. JHJ and GH were principal investigators and involved in the study design, data collection, and interpretation of the results. PK and IAvZ wrote the article that was critically revised by all coauthors.
DISCLOSURESThe authors have no conflicts of interest to disclose.
SOURCES OF FUNDINGThis work was supported by the MDS-RIGHT project, which has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement 634789. GG was supported by NIH grants R01 MH104964 and R01 MH123451. AOdG and JHJ were supported by a grant from the Dutch Cancer Society (grant number 10813). The Lifelines initiative has been made possible by subsidy from the Dutch Ministry of Health, Welfare and Sport, the Dutch Ministry of Economic Affairs, the University Medical Center Groningen (UMCG), Groningen University and the Provinces in the North of the Netherlands (Drenthe, Friesland, Groningen).
REFERENCES 1. Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371:2488–2498. 2. Thompson DJ, Genovese G, Halvardson J, et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature. 2019;575:652–657. 3. Loftfield E, Zhou W, Graubard BI, et al. Predictors of mosaic chromosome Y loss and associations with mortality in the UK Biobank. Sci Rep. 2018;8:12316. 4. Forsberg LA, Rasi C, Malmqvist N, et al. Mosaic loss of chromosome Y in peripheral blood is associated with shorter survival and higher risk of cancer. Nat Genet. 2014;46:624–628. 5. Zink F, Stacey SN, Norddahl GL, et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130:742–752. 6. Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121. 7. Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371:2477–2487. 8. Ljungström V, Mattisson J, Halvardson J, et al. Loss of Y and clonal hematopoiesis in blood-two sides of the same coin? Leukemia. 2021;36:889–891. 9. Ouseph MM, Hasserjian RP, Dal Cin P, et al. Genomic alterations in patients with somatic loss of the Y chromosome as the sole cytogenetic finding in bone marrow cells. Haematologica. 2021;106:555–564. 10. Stolk RP, Rosmalen JG, Postma DS, et al. Universal risk factors for multifactorial diseases: lifelines: a three-generation population-based study. Eur J Epidemiol. 2008;23:67–74. 11. Scholtens S, Smidt N, Swertz MA, et al. Cohort profile: lifelines, a three-generation cohort study and biobank. Int J Epidemiol. 2015;44:1172–1180. 12. van Zeventer IA, de Graaf AO, Wouters H, et al. Mutational spectrum and dynamics of clonal hematopoiesis in anemia of older individuals. Blood. 2020;135:1161–1170. 13. Kamphuis P, van Bergen M, van Zeventer IA, et al. Abnormal platelet counts and clonal hematopoiesis in the general population. Hemasphere. 2023;7:e821. 14. van Zeventer IA, de Graaf AO, Koorenhof-Scheele TN, et al. Monocytosis and its association with clonal hematopoiesis in community-dwelling individuals. Blood Adv. 2022;6:4174–4184. 15. Loh PR, Genovese G, Handsaker RE, et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature. 2018;559:350–355. 16. Loh PR, Genovese G, McCarroll SA. Monogenic and polygenic inheritance become instruments for clonal selection. Nature. 2020;584:136–141. 17. van Zeventer IA, Salzbrunn JB, de Graaf AO, et al. Prevalence, predictors, and outcomes of clonal hematopoiesis in individuals aged ≥80 years. Blood Adv. 2021;5:2115–2122. 18. Saiki R, Momozawa Y, Nannya Y, et al. Combined landscape of single-nucleotide variants and copy number alterations in clonal hematopoiesis. Nat Med. 2021;27:1239–1249.
留言 (0)