
Remember me
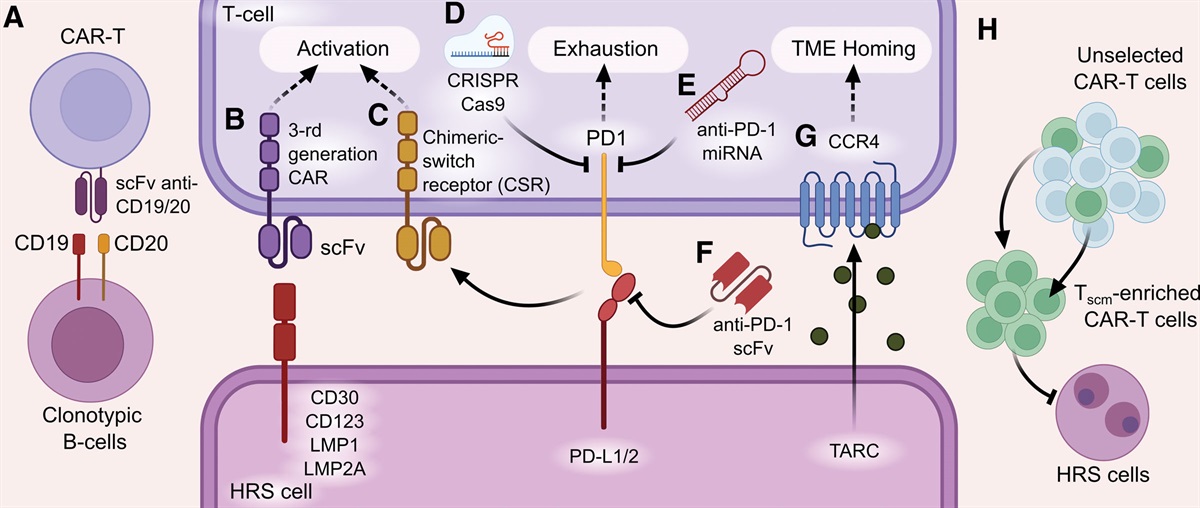


T cells engineered to express a chimeric antigen receptor (CAR) are undoubtedly among the most promising therapeutic modalities in oncology, and particularly in hematology. The first evidence of successful in vivo expansion, antitumor activity, and long-term persistence of a second-generation anti-CD19 CAR T-cell product came from a patient with a relapsed form of chronic lymphocytic leukemia (CLL) who during the course of the disease had acquired 17p deletion and had become refractory to third-line chemoimmunotherapy.1 Since then, several advances have been achieved in the field of genetically modified T-cell immunotherapy for lymphoid malignancies. These have led to the approval of 4 second-generation anti-CD19 CAR T-cell products, with indications that are covering an expanding range of diseases, including acute lymphoblastic leukemia,2,3 large B-cell lymphoma (BCL),4–6 mantle cell lymphoma,7,8 and follicular lymphoma.9,10 Despite the promising preclinical and early clinical results and the meaningful progress in the field, the clinical development of CAR T cells has been more challenging in the context of CLL due to some disease-specific critical issues.
The first limitation to the clinical applicability of CAR T cells in CLL is the patients’ population that mainly consists of elderly and comorbid subjects.11 Still, although a high level of concern about a lower age–related tolerability is justified, evidences from the pivotal ZUMA-1 trial and from early real-world experiences indicate that CAR T-cell therapy can be delivered to older patients with comorbidities, at least in the more explored setting of large BCLs.12–15
The second critical issue for the development of CAR T-cell therapy for CLL is the significant expansion of the treatment options that has occurred in the last years, which may limit the clinical need for cellular immunotherapy. Since the early 2000, the addition of anti-CD20 monoclonal antibodies to chemotherapy has shown the potential to achieve prolonged remissions and significant improvement in overall survival (OS) compared to chemotherapy alone, especially in previously untreated patients without relevant comorbidities and presenting a disease with low-risk biological features.16,17 More recently, the clinical testing of Bruton tyrosine kinase (BTK) inhibitors and of the BCL2 protein inhibitor venetoclax, variably administered in sequence and/or in combination and in presence or absence of anti-CD20 monoclonal antibodies, has provided remarkable clinical results and superiority compared to chemoimmunotherapy for the treatment of all risk groups.18–23 That notwithstanding, targeted agents have some important limitations, which affect their curative potential, such as (1) the loss of efficacy and possible development of drug resistance mechanisms, frequently reported for continuous treatment regimens,24–27 and (2) the inferior efficacy observed in high-risk subtypes (such as those carrying TP53 aberrations or multiple cytogenetic abnormalities), in particular in the context of fixed-duration therapy. Furthermore, the use of targeted agents has not proven to be particularly beneficial for those cases of CLL that have transformed into an aggressive lymphoma (Richter syndrome), a clinical situation where treatment options are still very limited and prognosis remains definitely poor, with a median survival of less than 1 year.28 Accordingly, for patients who have failed treatment with BTK inhibitors and venetoclax, or who develop Richter syndrome, the identification of immune-based therapeutic strategies is worth investigation.
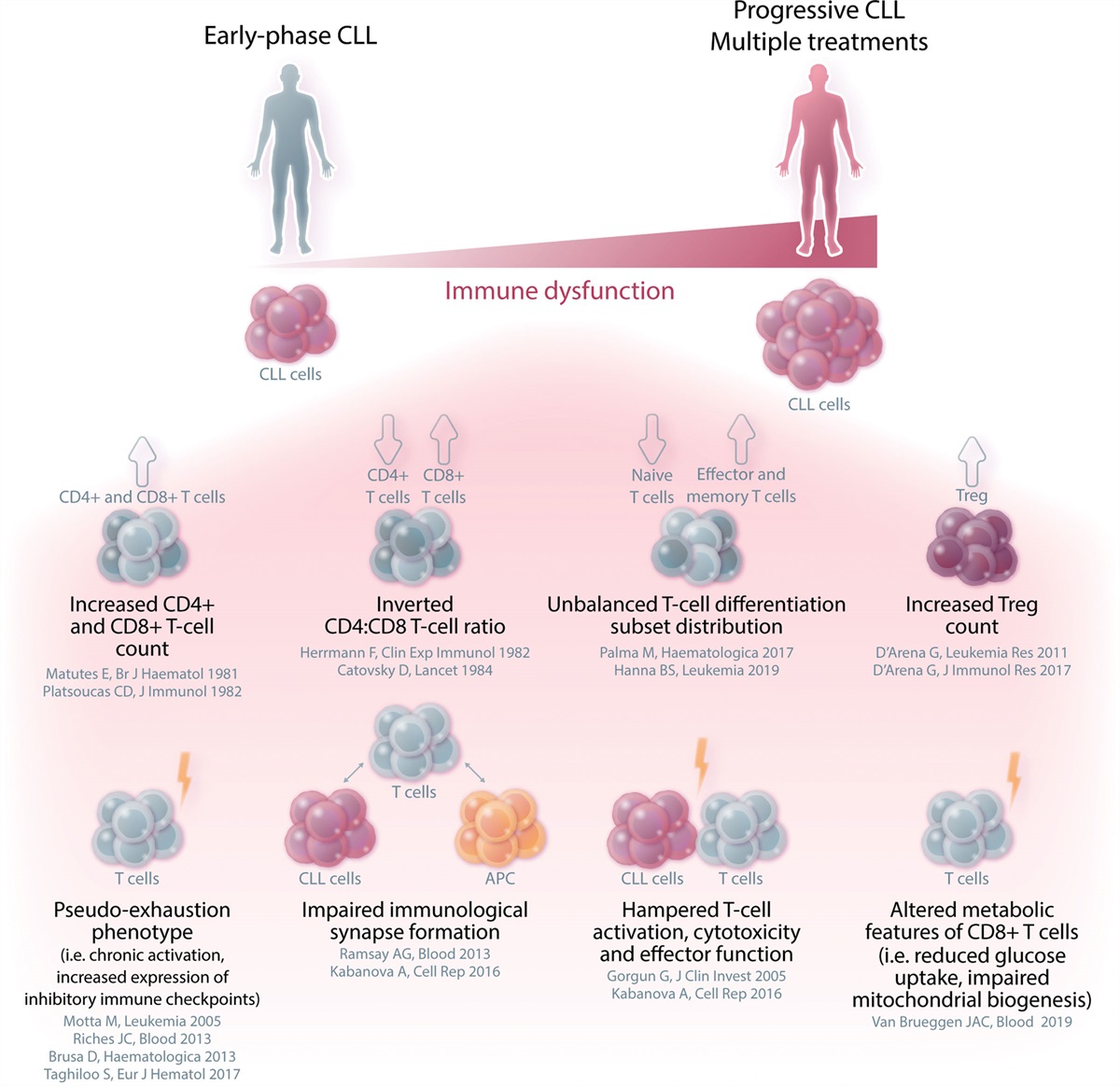
The third and most likely the main issue that has limited the development of successful CAR T-cell therapies for CLL is the difficulty in producing effective CAR T-cell formulations. This is mainly due to the intrinsic immunologic deterioration that characterizes the disease.29,30 Indeed differently from other hematological disorders, where the acquisition of antigen (Ag)-negative tumor variants represents a relevant cause of CAR T-cell therapy failure,31–33 in CLL the main reasons for treatment resistance can be found more likely in the production of defective or shortly persisting CAR T cells that are not effective in eradicating the tumor cells and therefore favor the development of Ag-positive relapses. CLL is characterized by a wide range of deep immune alterations and complex intrinsic T-cell defects, which already occur in the very early phases of the disease—at some degree even in the preneoplastic phase of monoclonal B-cell lymphocytosis34—and then progressively accumulate, in parallel with the increase in tumor burden and under the pressure of multiple lines of treatment (Figure 1). From the clinical standpoint, the relevance of these immunologic alterations is demonstrated by some manifestations that frequently occur during the disease course and are considered a hallmark of CLL. These include the increased susceptibility to infections, the lower response to prophylactic vaccinations, the higher risk of second primary malignancies, and the high frequency of autoimmune phenomena.35 Specifically, several T-cell alterations have been reported in CLL, such as the presence of an inverted CD4:CD8 T-cell ratio,36–38 the unbalanced differentiation subset distribution,39–42 the higher number of regulatory T cells (Tregs),43–47 and the increased expression of pseudoexhaustion markers and transmembrane inhibitory receptors, including CTLA-4, PD-1, TIM-3, or LAG-3.39–41,48–56 All of these T-cell alterations translate in functional impairments, including defects in immune synapse formation and in cytotoxicity.57–61 This subversion of T-cell–related immunity results in altered features of CLL-derived CAR T cells, which often present expression of exhaustion markers that cause reduced cytokine secretion, limited expansion capacity, and consequently, poor antitumor function.62 Of note, the manufacturing strategies developed up to now are not able to fully overcome the basic immunological alterations related to the disease and might even contribute to the acquisition of additional features of functional exhaustion and short in vivo persistence.
 Figure 1.:
Figure 1.: Immune system dysfunctions that characterize patients with CLL and can impact on CAR T-cell functionality. Phenotypic alterations and functional impairment characterize the T-cell compartment of CLL patients. Immune dysregulation is often present since the early stages of CLL, and exacerbates with disease progression and as a consequence of multiple lines of treatment, thus hampering immune competence. CAR = chimeric antigen receptor; CLL = chronic lymphocytic leukemia.
In this review, we provide a comprehensive overview of the clinical results achieved by CAR T-cell therapy in CLL and of the identified reasons, to date, of suboptimal efficacy. We specifically focus on immune-related mechanisms that contribute to the poor functionality and to the incomplete eradication or reappearance of Ag-positive leukemic cells. In addition, we describe strategies that aim to overcome the T-cell–related defects as well as strategies that are currently pursued to improve the clinical efficacy of CAR-modified cellular therapies. These efforts should lead to the development of novel cellular therapies capable of counteracting the more aggressive and still incurable forms of the disease.
SECOND-GENERATION ANTI-CD19 CAR T CELLS FOR THE TREATMENT OF CLL: MORE THAN 10 YEARS OF ATTEMPTSSecond-generation anti-CD19 CAR T-cell constructs—comprising an Ag-binding portion, a transmembrane domain, an intracellular signaling domain of CD3ζ chain, and a single costimulatory domain2,6,63–66—were the first to be broadly evaluated in the clinical settings and to be approved for use in the clinical practice. Patients with CLL were included in the heterogeneous patient populations enrolled in early studies exploring the safety and efficacy of second-generation anti-CD19 CAR T-cell products, but later clinical trials have mainly focused on aggressive BCLs. As of December 2023, no CAR T-cell product has been approved for the treatment of patients with CLL. A summary of the efficacy and safety results achieved in CLL patients with anti-CD19 CAR T-cell products is presented in Table 1.
Table 1 - Summary of Results Obtained in Patients with CLL Using Anti-CD19 CAR T Cells Product Administered Product Specifics Patients Evaluable for Response, Number Lymphodepleting Chemotherapy ORR CR Rate Median DOR Median PFS Median OS Safety Reference Axi-cel (KTE-C19) + IV IL-2 Second generation CLL = 4 Flu/Cy 75% (3/4) 25% (1/4) 7 months NA NA Grade ≥3 AEs 100% (4/4) 67 Axi-cel (KTE-C19) Second generation CLL = 4a Including patients from the previous series.
b Data available for the whole cohort of the patients enrolled in the study.
c Data available for the NHL/CLL cohort of the patients enrolled in the study.
AEs = adverse events; Axi-cel = axicabtagene ciloleucel; Brexu-cel = brexucabtagene autoleucel; CAR = chimeric antigen receptor; CLL = chronic lymphocytic leukemia; CR = complete response; CRS = cytokine release syndrome; Cy = cyclophosphamide; DOR = duration of response; Flu = fludarabine; HD = high dose; IV = intravenous; LD = low dose; Liso-cel = lisocabtagene maraleucel; MSKCC = Memorial Sloan Kettering Cancer Center; NA = not available; NE = neurologic events; NHL = non-Hodgkin lymphoma; NR = not reached; ORR = overall response rate; OS = overall survival; PFS = progression-free survival; RS = Richter syndrome; Tisa-cel = tisagenlecleucel.
In 2012, a group at the National Cancer Institute published the results obtained in 8 patients treated with their anti-CD19 CAR T cells (later called axicabtagene ciloleucel [axi-cel]) combined with a course of intravenous interleukin (IL)-2.67 Four patients with extensively pretreated CLL (median number of prior therapies, 4.5) were included, among whom 3 had a response, including one who had a prolonged complete remission (CR, >15 months). A modified treatment plan that omitted IL-2 administration was later applied for the treatment of 4 additional patients with CLL, who all had a response with a CR rate of 75%.68 At the last update presented in 2020, a persistent response of more than 3 years was reported in 4 of 8 patients with CLL, among whom 2 had a response lasting more than 6 years.69
A similar CAR T-cell product (brexucabtagene autoleucel [brexu-cel])—which differs from axi-cel only in the slightly different manufacturing process of removing circulating CD19-expressing malignant cells to avoid the possible activation and exhaustion of anti-CD19 CAR T cells during the production—is currently under evaluation for the treatment of relapsed/refractory CLL within the phase I ZUMA-8 study. Initial results from 15 patients, of whom 80% previously received >3 treatment lines, were recently presented, with an overall response rate (ORR) of 47% and a CR rate of 13%.70 Cytokine release syndrome (CRS) developed in 12 of 15 patients but was only grade ≥3 in 1 patient, whereas all-grade and grade ≥3 neurotoxicity developed in 11 of 15 and 3 of 15 patients, respectively. Overall, the safety and preliminary efficacy results of this product appear limited, suggesting the need for further optimization.
Tisagenlecleucel (tisa-cel) was also evaluated for the treatment of patients with CLL: the tumor regression observed in the first patient with CLL was reported in 2011,1 followed by preliminary results obtained in 3 patients, all of whom responded to the therapy.71 Mature results from an expanded cohort of patients with relapsed/refractory CLL who received a median of 5 previous therapies were later presented (n = 14).72 The ORR was 57%, and in 29% of the patients a CR with undetectable minimal residual disease (MRD) was reported. A persistent response was achieved in patients who obtained a CR (median duration of response [DOR], 40 months) with no relapses noted during the follow-up, whereas for patients who obtained a partial response (PR), median DOR was only 7 months. Nine of 14 patients developed CRS, which was grade 3-4 in 6 patients, and associated with tisa-cel peak expansion and clinical response.
Based on these encouraging results, and with the aim of determining an optimal cell dose for future application, the authors then performed a randomized, phase II study in patients with relapsed/refractory CLL.73 Forty-two previously treated patients with CLL (median number of prior therapies, 3.5) were randomly assigned to receive 2 different doses of tisa-cel (5 × 108 or 5 × 107 CAR T cells). Thirty-eight patients received the treatment and were evaluable for safety, and 32 were evaluable for response. Despite the higher response rates achieved with high dose compared to low dose (ORR, 53% versus 31%; CR rate, 36% versus 15%), there were no differences in progression-free survival (PFS) and OS between the cohorts. Of note, PFS and OS were significantly longer in patients who achieved a CR versus those who did not (median PFS, 40 months versus 1 month, P < 0.0001; median OS not reached versus 64 months, P = 0.035), regardless of the dose received. Toxicity was similar in the 2 dose groups: 63% of patients overall developed all-grade CRS and 24% developed grade 3-4 CRS.
Interestingly, investigators from the University of Pennsylvania have recently reported on the long-lasting persistence of circulating CAR T cells, together with sustained disease remission, in 2 of the patients with CLL who were treated with tisa-cel and achieved a CR in 2010,74 thus providing the proof of concept evidence that CAR T cells may potentially be considered curative for this disease.
An additional second-generation anti-CD19 CAR T-cell construct was developed by a group at Memorial Sloan Kettering Cancer Center. Initially reported data showed no response in 8 patients with purine analog refractory CLL.75 The protocol was later modified, optimizing the conditioning regimen by adding fludarabine (Flu) and permitting the inclusion of ibrutinib pretreated patients. Sixteen patients with relapsed or refractory CLL were treated (median number of prior therapies, 4) and an objective response was observed in 6 of 16 (38%) CLL patients. Three of 12 (25%) evaluable patients with CLL who received conditioning chemotherapy achieved CR by International Workshop on Chronic Lymphocytic Leukemia (IWCLL) criteria.76 Alongside the unsatisfactory efficacy, toxicity of this CAR T-cell formulation was relevant. Indeed, all 16 patients experienced CRS, although only 2 had a grade ≥3 event.
As it emerges from data presented to date, the clinical efficacy of second-generation CAR T cells has been initially disappointing in CLL, despite the nonnegligible toxicities, thus prompting investigation of the immunological and biological mechanisms responsible for the limited functionality and designing novel protocols of production and administration to achieve successful CAR T-cell therapies.
REASONS FOR SUBOPTIMAL EFFICACY AND DETERMINANTS OF CAR T-CELL FUNCTIONALITY IN CLLOver time, several groups have investigated the possible reasons underlying the suboptimal clinical efficacy obtained with second-generation anti-CD19 CAR T-cell products in CLL, focusing on the identification of T-cell–related determinants capable to predict CAR T-cell functionality.
One of the main factors affecting the clinical efficacy of CAR T cells is the cellular composition of the infused genetically modified T lymphocytes. Reported data show that the ability of CAR T cells to expand in vivo positively correlates with the frequency, within the infused product, of a CAR-expressing CD8+CD45RA+CCR7+ T-cell population: a cell subset phenotypically close to T memory stem cells (TSCM), which have self-renewal capacity and the ability to rapidly differentiate into effector T cells upon Ag exposure.77 However, in this study, no patient achieved a sustained clinical response, thus precluding the possibility to correlate the composition and phenotype of CAR T cells with the clinical outcome.
A further demonstration of the importance of the subset composition of the administered cellular products was provided by Sommermeyer et al, who were able to recognize the enhanced potency of anti-CD19 CAR T cells composed of defined T-cell subsets compared with those produced from unselected peripheral blood mononuclear cells (PBMCs) obtained from patients with BCL.78 Specifically, individual CD8+ and CD4+ T-cell subsets from both patients with B-cell malignancies and from normal donors were purified and used to produce CAR T cells, whose functional activity was then assessed in vitro and in vivo. Overall, CAR T cells derived from less-differentiated naive (TN) and central memory (TCM) CD4+ or CD8+ T cells were functionally more effective in terms of cytokine production than those derived from more differentiated effector memory (TEM) CD4+ or CD8+ T cells. In addition, in vivo experiments show that a third-generation CAR T-cell product containing the CD28 and 4-1BB costimulatory domains administered to immunodeficient mice (nonobese diabetic/severe combined immunodeficiency/γc−/−) engrafted with a CD19+ Burkitt lymphoma–derived cell line was more potent in eradicating the tumor when administered as a formulation containing a 1:1 ratio of CD8+ CAR T cells derived from TCM with CD4+ CAR T cells, particularly if derived from the TN subset. Based on these observations, one possible reason for the reported heterogeneous efficacy of CAR T-cell therapy in patients with lymphoproliferative disorders could be found in the patient-to-patient variability in terms of subset distribution within the CAR T-cell population produced from unselected T-cell sources. Since the quality of the CAR T-cell product strictly depends on the characteristics of the initial T-cell population, it is important to take into account that CLL patients have an underrepresented compartment of circulating TN cells, which are also characterized by a limited expansion potential—2 hallmarks that may relevantly challenge the generation of an effective CAR T-cell population.79
Precisely with the purpose of recognizing parameters predictive of CAR T-cell functionality, Fraietta et al performed an extensive analysis evaluating several T-cell–related features in a large cohort of 41 patients with CLL treated with 4-1BB-based anti-CD19 CAR T cells (CTL019).62 Clinical efficacy was not predicted by patient and disease characteristics (ie, tumor burden, presence of TP53 gene aberration, and number of previous therapies), whereas parameters predictive of successful treatment could be identified both at the level of the premanufacturing T cells and of the infused CAR T cells. Results from this analysis showed that premanufacturing unmanipulated T cells from nonresponding patients display upregulation in genes involved in T-cell exhaustion (ie, DUSP4, CXCL13), activation (ie, IL1A, STAT3), glycolysis (ie, B4GALNT1, DNAJC12), and apoptosis (ie, EMP1, DRAM1).62 Additionally, leukapheresis products from responding patients were characterized by an elevated frequency of CD8+ TSCM cells, thus confirming that the cellular composition of the initial T-cell source also has an impact on the functional properties of the administered cellular product. In line with these observations also in other hematologic and solid tumors,80,81 both the presence of specific T-cell subsets and the phenotypical and functional characteristics of the premanufacturing T cells were identified as informative parameters of the functionality of the infused CAR T cells. In this context, a point that is still a matter of debate is the impact of previous chemoimmunotherapy, which can certainly affect the initial composition and fitness of the T-cell compartment, and the possibility to effectively generate fully functional CAR T cells.82
In their comprehensive analyses, Fraietta et al have also strengthened the concept that the characteristics of the final CAR T-cell formulation have a relevant impact on patients’ clinical outcome. They observed that in CAR T-cell–treated patients, the therapeutic efficacy was primarily associated with parameters of preserved functionality at the level of the infused CAR T cells, as defined by their in vivo expansion and persistence. In addition, CAR T cells from responding versus nonresponding patients were characterized by a different transcriptomic profile (early memory differentiation versus late memory/effector T-cell differentiation, respectively) and by a decreased dependency from aerobic glycolysis. The impact of the metabolic asset on the functionality of CAR T cells was also confirmed by van Bruggen et al, who demonstrated that patients with CLL achieving a CR after treatment with CTL019 have a higher CAR T-cell mitochondrial mass compared with nonresponder patients.83
Another factor that has shown to exert a relevant impact on the clinical outcome is the expression profile of inhibitory immune checkpoints on the infused CAR T cells. Kong et al reported significantly higher levels of CTLA-4 and TIM-3 or LAG-3 expression on CAR T cells of nonresponding patients compared with responding patients.84 Moreover, in nonresponding patients, at the peak of the in vivo expansion phase, a higher proportion of CAR T cells coexpressing PD-1 and TIM-3 was observed, and the upregulation of these markers of exhaustion was paralleled by the acquisition of defects in expansion capacity and in perforin-mediated cytotoxicity.
Overall, these observations highlight the meaningful impact of the composition and quality of both the premanufacturing T-cell source and the final CAR-modified T-cell products in determining the postinfusion expansion, the in vivo persistence, and, ultimately, the antitumor functions and clinical efficacy of CAR T cells.
STRATEGIES TO IMPROVE CAR T-CELL EFFICACY IN CLL: OVERCOMING T-CELL DYSFUNCTIONSGiven the disappointing clinical efficacy achieved in CLL by the same anti-CD19 CAR T-cell formulations that are instead successful in other lymphoid malignancies,10,13,85 different strategies have been tested and are currently under investigation with the aim of reversing CLL-related immunological dysfunctions and obtaining a cellular product with more powerful antitumor functions (Figure 2).
 Figure 2.:
Figure 2.: Overview of strategies to improve CAR T-cell efficacy in CLL through the targeting of disease-related immune dysfunctions. The steps for the generation of autologous CAR T cells from a CLL patient are depicted. Several strategies are currently under investigation with the aim to overcome T-cell alterations and to obtain CAR T-cell products with improved antitumor efficacy in CLL. CAR = chimeric antigen receptor; CLL = chronic lymphocytic leukemia.
Improving the composition of infused CAR T-cell productsBased on previously cited preclinical data proving the key role of the T-cell subset composition on CAR T-cell efficacy,78 a group at Fred Hutchinson Cancer Research Center focused their work on the development of an optimized second-generation anti-CD19 CAR T-cell formulation consisting of a precise dose of CD4+ and CD8+ CAR T cells. JCAR014 is manufactured from separate subsets of CD4+ T cells and either bulk CD8+ T cells or CD8+ TCM cells, and formulated for infusion in a 1:1 ratio of CD4+:CD8+ CAR T cells. This product was studied in a phase I/II clinical trial that enrolled 24 heavily pretreated and high-risk patients with aggressive CLL or Richter transformation (n = 19 and n = 5, respectively).86 Of note, all patients had received previous ibrutinib therapy (median duration of ibrutinib administration, 13 months) and most of them had experienced ibrutinib failure. CAR T cells were administered after lymphodepleting chemotherapy consisting of cyclophosphamide (Cy), Flu, or Cy plus Flu. In the whole cohort, 16 patients responded (ORR, 70%) as per IWCLL imaging criteria assessed by the investigators, with a CR rate of 17% at 4 weeks after CAR T-cell infusion. Among patients who received Cy plus Flu lymphodepletion and <2 × 106 CAR T cells/kg, 88% reached undetectable MRD by flow cytometry in the bone marrow 4 weeks after CAR T-cell administration, and 58% of them also had molecularly undetectable MRD (assessed by immunoglobulin heavy chain [IGH] sequence). At a median follow-up of 6.6 months, estimated median PFS was 8.5 months and median OS was not reached. The response to treatment correlated with outcome since patients who achieved either a CR or PR had longer PFS and OS compared with those who experienced treatment failure (CR: median PFS 9.8 months, median OS not reached; PR: median PFS not reached, median OS not reached; treatment failure: median PFS 1.1 months, median OS 11.2 months). Of note, in this cohort, MRD clearance by IGH sequencing was predictive of longer PFS, independently of the response achieved by IWCLL criteria. Regarding treatment-related toxicities, despite the high frequency of occurrence (83%), CRS presented as grade 1-2 in most patients (75%), with only 1 patient experiencing grade 4 and 1 experiencing grade 5 CRS. Eight of 24 patients (33%) had neurotoxicity (grade 1-2: 2 patients; grade 3: 5 patients; grade 5: 1 patient). Although the incidence of serious CRS was low, neurotoxicity could represent a limit for this CAR T-cell product, especially considering that patients with CLL are frequently old and with meaningful comorbidities.
Lisocabtagene maraleucel (liso-cel, JCAR017) is a more recently introduced CAR T-cell product that uses the same construct as JCAR014 and maintains a fixed 1:1 ratio of CD4+:CD8+ CAR T cells, but differs in the manufacturing process, because (1) cytokines instead of Ag-presenting cells are used during the ex vivo T-cell stimulation phase87 and (2) the early selection phase of CD4+ and CD8+ T cells involves discharging from the leukapheresis the non-T-cell elements to limit the risk of transducing leukemic cells with the anti-CD19 CAR and to favor a more efficient expansion of CAR T cells.87,88 JCAR017 is currently under evaluation for the treatment of relapsed/refractory CLL in the phase I/II TRANSCEND CLL 004 trial, whose primary efficacy analysis results were recently published.89 In this study, 137 patients were enrolled, 117 of whom received liso-cel. Most of the patients (83%) were considered high-risk, carrying a TP53 alteration and/or unmutated IGHV genes and/or complex karyotype. Notably, all treated patients had received previous ibrutinib treatment, with most of the patients developing resistance to BTK inhibitor (88%) and a consistent subset showing failure or disease recurrence upon venetoclax-based therapy (60%). Patients received liso-cel at 2 different dose levels (ie, 50 × 106 CAR T cells [n = 9] or 100 × 106 CAR T cells [n = 108]) and 96 of them were evaluable for efficacy: 46 achieved a response (ORR, 48%) and 17 achieved a CR (18%). These response rates were comparable to those obtained in the subcohorts of patients who progressed after BTK inhibitors and failed venetoclax therapy (ORR, 43%; CR, 18%). At a median follow-up of 21 months, the median PFS was 17.8 months and the median DOR was 35.2 months, with median DOR for CR patients not reached. Among 49 patients included in the primary efficacy analysis set and treated with the higher dose level, at any time during follow-up, 63% and 59% achieved an undetectable MRD in the blood and in the bone marrow, respectively, as assessed by next-generation sequencing. The concordance of detectable and undetectable MRD between blood and bone marrow was 96%, and all patients who responded to the therapy reached undetectable MRD in both compartments. Most of the adverse events were low grade. Among 117 patients evaluated for safety, 99 (85%) had CRS (grade 1-2: 76%; grade 3: 9%) and 53 patients (45%) had neurological events (grade 1-2: 26%; grade 3: 18%; grade 4: 1%). Forty-three patients died after CAR T-cell infusion: 27 due to progressive disease, 5 due to adverse events occurring within 90 days of CAR T-cell infus
Comments (0)