
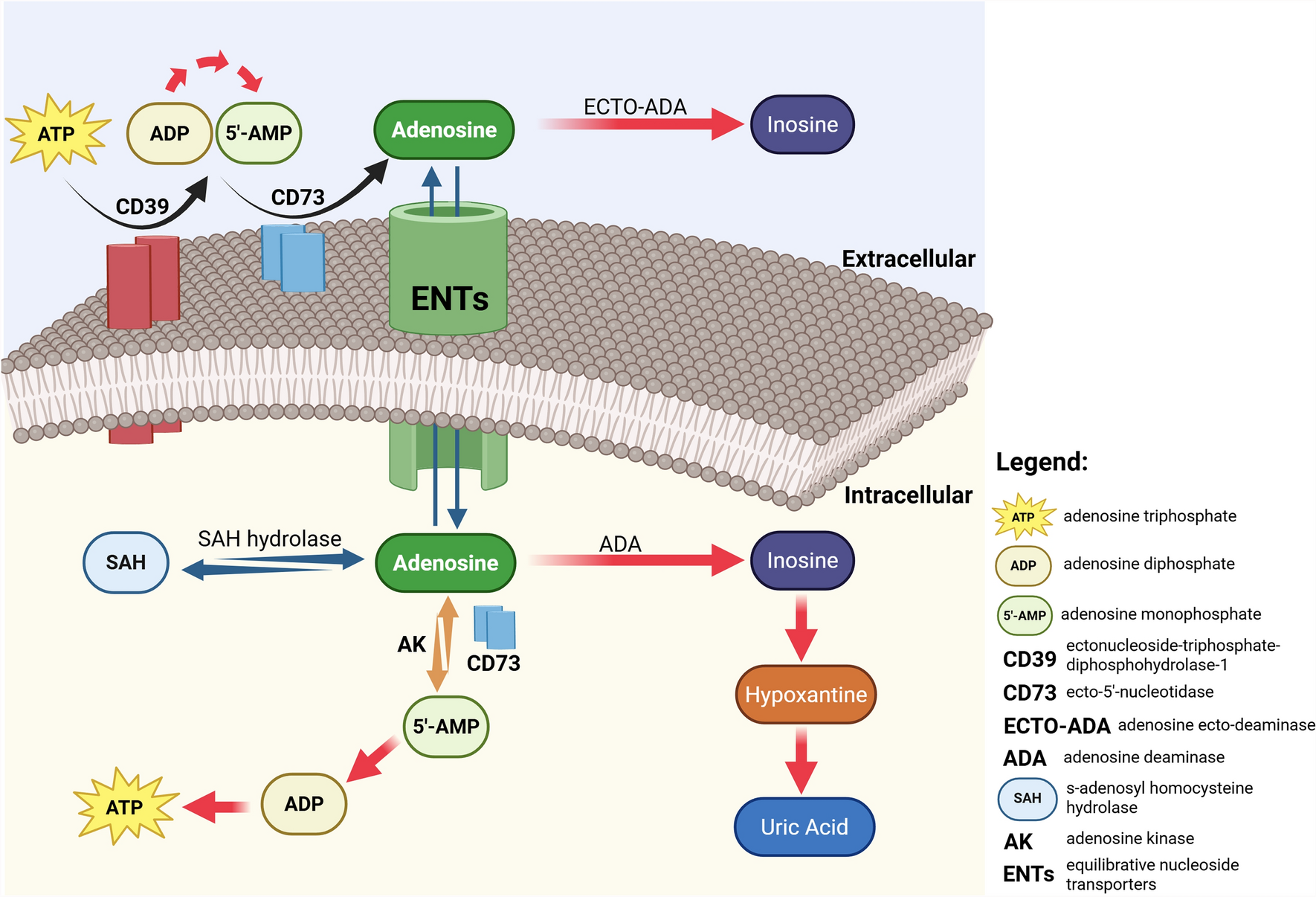
Remember me



Like other organelles, mitochondria can also be released by mammalian cells. The mitochondria possess a number of characteristics that facilitates their detection within EVs or the extracellular space. Mitochondria exhibit a distinct membrane topology, making them recognizable under electron microscopy even when fragmented. In addition, mitochondria possess their own DNA, allowing for another handle that can be used for detection in fluorescence imaging or biochemical-based analyses. These properties have allowed the discovery of cellular settings that display mitochondrial extracellular release. The released content can be functional mitochondria or bio-active mitochondrial components for modulating nearby cells. Emerging data also suggest that damaged mitochondria can be selectively picked out and shipped to the extracellular space, presumably for quality control.
Mito-release for immune-activationCells release mitochondria or mitochondria-containing EVs to evoke an immune response in target cells [27,28,29,30,31]. For example, monocytic cells release free mitochondria and microvesicles with mitochondrial contents upon lipopolysaccharide (LPS) stimulation [30]. These released mitochondria and microvesicles induce type I IFN (interferon) and TNF (tumor necrosis factor) dependent responses, including the induction of ICAM-1 (intercellular adhesion molecule), VCAM (vascular cell adhesion molecule) mRNA expression level as well as IL-8 production in endothelial cells [30]. Furthermore, activated platelets could release mitochondria, again either as free organelles or within membrane-encapsulated microparticles, into the blood plasma [27]. Blood plasma sPLA2-IIA (Secreted phospholipase A2 group IIA), an endogenous phospholipase specific for bacteria, then hydrolyzes lipids on the mitochondrial membrane to release various lysophospholipids, free fatty acids, and mtDNA to promote leukocyte activation [27]. It has also been shown that activated microglia release fragmented, dysfunctional mitochondria to activate astrocytes, leading to the propagation of neuroinflammation [28].
That mitochondrial release can lead to immune modulation is thought to arise from proinflammatory signals that reside within mitochondria. Unlike nuclear DNA, the mitochondrial genome is circular, free of histones, and shows limited methylation on CpG islands. These characteristics of mitochondrial DNA share resemblances with the bacterial genome, which act as potential agonists of immune response once present in the cytosol or the extracellular environment [32]. Aside from mtDNA, mitochondrial components such as N-formyl peptides, cardiolipin, and cytochrome C also display proinflammatory properties.
Mito-release for boosting mitochondrial functions in recipient cellsCan the extracellular release of mitochondria, or components thereof, improve mitochondrial functions in recipient cells? Can the released material even be functional, and capable of integrating into the existing mitochondrial network of the recipient cells upon internalization?
Both neuronal cells and mesenchymal stem cells (MSCs) have been found to release EVs containing functional mitochondria [33,34,35,36,37]. Neuronal cells release mitochondria with intact ultrastructure and membrane potential, which have been shown to alleviate stress in recipient cells [33, 34]. Similarly, MSCs have also been observed to release functional mitochondria into the extracellular space, as demonstrated by co-culture studies and mitochondrial membrane potential-dependent dye staining [35,36,37]. Upon uptake by recipient cells, these MSC-released mitochondria can fuse with existing mitochondrial networks, indicating their functional capacity [37]. This transfer of functional mitochondria has been linked to enhanced bioenergetics, as evidenced by increased oxidative phosphorylation and ATP production in recipient cells [35,36,37]. These properties may offer a potential explanation for the observed health benefits associated with MSC-based therapies.
Mito-release for quality controlMitochondrial release during cell migrationCells undergoing migration can generate membrane protrusions and release EVs known as migrasomes, which contain cellular components that accumulate and pinch off from the tip of the protrusions [38]. Studies indicate that migrasomes are oftentimes assembled within membrane microdomains enriched with tetraspanins and cholesterol. The presence of tetraspanins and cholesterol on these microdomains is believed to enhance membrane rigidity, facilitating the formation of a bulging membrane structure necessary for migrasome development [39].
Recent studies have shown that when migrating cells are treated with low concentrations of the oxidative phosphorylation uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP at 2 µM), dysfunctional mitochondria can be released into the extracellular space through migrasomes (Fig. 2a). The mitochondria residing in these migrasomes display a swollen morphology, generate higher than normal levels of reactive oxygen species, and possess lower mitochondrial membrane potential when compared to mitochondria in the cytoplasm [40]. Mechanistically, it has been found that migrasome-released mitochondria recruit less of the motor protein dynein but more KIF5B, leading to their selective accumulation at the cell periphery for extracellular release. This selective release of dysfunctional mitochondria suggests that mitocytosis (a term used to describe the migrasome-mediated mitochondria release) is a quality control mechanism for mitochondria. Studies have demonstrated that mitocytosis is required to maintain mitochondrial respiration and membrane potential in neutrophils and is necessary for macrophage differentiation in vivo. Interestingly, higher concentrations of CCCP (10 µM) lead to intracellular degradation of mitochondria through mitophagy instead of mitocytosis. Future studies will better define the interplay between mitocytosis and mitophagy in the quality control of mitochondria in migrating cells.
Fig. 2
Mitochondria Extracellular Release as a Quality Control Mechanism. a. Damaged mitochondria are selectively transported to migrasomes through retraction fibers and released. b. (1) Neurons are able to deliver damaged mitochondria to adjacent astrocytes for their degradation. (2) Meanwhile, astrocytes can deliver functional mitochondria back to neurons in order to compensate for malfunctioning mitochondria. (3) When neurons suffer from undesirable conditions, Schwann cells can release mtDNA and cytochrome C as warnings to inform neurons of potential danger and degeneration. c. Mitochondria quality control in cardiomyocytes occurs through two pathways. One, cardiomyocytes can release damaged mitochondria to cardiac macrophage through exophers for degradation. Two, the damaged mitochondria can also be degraded by mitophagy. Figure created with BioRender.com
Horizontal mitochondrial transfer in the neuronal systemNumerous studies indicate that neuronal mitochondria can be transferred into glial cells [41,42,43,44,45]. Some of these studies suggest that EVs facilitate this mitochondrial delivery. For instance, 3D images of mouse retinal ganglion cell axons obtained through serial block-face scanning electron microscopy reveal that axonal mitochondria can be sent into the extracellular space through the formation of protrusions [45]. Furthermore, when retinal ganglion cell mitochondria were labeled with the tandem-fluorescent mitochondrial marker MitoEGFPmCherry, it was discovered that some of these mitochondria are eventually targeted to the lysosomes of nearby astrocytes. Subsequent examination using the TUNEL assay revealed that the mitochondrial DNA from these expelled mitochondria undergoes degradation within astrocytes. Collectively, these findings suggest that neuronal mitochondria can be released by axons and subsequently targeted to neighboring astrocytes for degradation. The presence of disease-promoting mutations in Optineurin, a known mitophagy-related protein, enhances this mitochondrial degradation activity [46]. This transcellular mitochondrial degradation activity has been termed "transmitophagy" (Fig. 2b).
The release of neuronal mitochondria can be amplified when challenged with various mitochondrial toxins. It is well-established that mitochondrial toxins can induce the release of mitochondrial components from neurons and affect nearby glial cells [47]. For instance, administration of 6-hydroxydopamine (6-OHDA) to mice leads to degeneration of nearby dopaminergic neurons, resulting in the shedding of micron-sized spherical particles (spheroids) from their axons [44]. These spheroids accumulate damaged mitochondria positive for PINK1/Parkin markers, which eventually enter the lysosomes of neighboring astrocytes. Similarly, employing optogenetic techniques to induce ROS-stress in mitochondria [48] within cone photoreceptor cells leads to the delivery of these ROS-stressed mitochondria into the lysosomes of Muller glia cells [42]. Electron microscopy-based analysis revealed that some of the mitochondria may have been transported from cone photoreceptors to Muller glia cells through budding of the plasma membrane. Both observations suggest that damaged mitochondria are released extracellularly and contribute to the occurrence of neuron-to-glia transmitophagy.
Conversely, it has also been demonstrated that astrocytes are capable of packaging mitochondria into EVs [34]. Electron microscopy studies have shown that cultured astrocytes release EVs containing mitochondria with intact cristae structures. These astrocyte-released mitochondria can be stained with mitochondria dyes that are dependent on membrane potential, indicating their functionality. When astrocytes are co-cultured with neuronal cells, the EVs released by astrocytes, containing functional mitochondria, can be taken up by neuronal cells and even fused into the neuronal mitochondrial network. Consequently, these EVs have been observed to enhance neuronal viability. For example, when astrocyte-released EVs containing functional mitochondria were administered into the brains of mice subjected to focal cerebral ischemia, they upregulated cell survival signals in mouse brain neurons. The observation that EVs can mediate neuron-to-glia transmitophagy of damaged mitochondria, as well as the delivery of functional mitochondria from glial cells back to neurons to improve neuron survival, suggests that EV-mediated mitochondrial exchange in the neuronal system may serve as a mitochondrial quality control mechanism.
Horizontal mitochondrial transfer in the heartResearch findings indicate that EV-mediated transmitophagy may play a role in facilitating mitochondrial quality control in the heart, similar to what was observed in the neuronal system (Fig. 2c). Studies have shown that the removal of cardiac-resident macrophages (cMacs) in mice leads to perturbations in mitochondrial qualities within cardiomyocytes. In the absence of cMacs, mitochondria in cardiomyocytes exhibit reduced cristae density and a decreased capacity for ATP production. Conversely, it has been observed that cardiomyocytes release micron-sized EVs, termed exophers, which contain structurally perturbed mitochondria. This process of EV-mediated mitochondrial disposal is upregulated when the mitochondria within mouse cardiomyocytes are subjected to stress, such as the administration of isoproterenol. Through the use of a pH-sensitive, mitochondria-targeted fluorescent reporter called mtKeima, researchers have determined that cardiomyocyte-derived extracellular mitochondria are engulfed by cMacs for lysosomal degradation. Taken together, these findings suggest that the cardiomyocyte-to-cMacs transmitophagy contributes to the quality control of mitochondria in the heart [49].
The ability of cMacs to remove damaged mitochondria from the heart implies a potential therapeutic value of these cells in mitigating heart-related disorders. Supporting this expectation, studies have demonstrated that the transplantation of cMacs into the pericardial space can prevent sepsis-induced cardiomyopathy in mouse models [50]. This beneficial effect is thought to be mediated by cMacs' ability to remove dysfunctional mitochondria from cardiomyocytes, thereby preserving cardiac function.
Horizontal mitochondrial transfer by the adipose tissueAmple observations indicate that adipocytes undergo EV-mediated release of oxidized mitochondrial components. When brown adipose tissue (BAT) is exposed to cold stress, a condition known to stimulate BAT mitochondrial ROS production to sustain thermogenesis [51, 52], there is a notable increase in the release of specific mitochondrial proteins. These proteins, such as the pyruvate dehydrogenase E1 subunit beta (PDHβ) and pyruvate decarboxylase, are transported via larger EVs (approximately 350 nm in diameter) [53]. Remarkably, the proteins released through these EVs exhibit signs of oxidation, including the presence of 4-Hydroxynonenal (4-HNE) and the formation of carbonyl protein adducts. Analysis of the released PDHβ reveals a higher degree of cysteine oxidation compared to its intracellular counterpart. Adipose tissue-resident macrophages play a crucial role in clearing these EVs induced by cold stress, thus preventing dysfunction in BAT.
In another scenario, mimicking obesity-associated impairment of mitochondria in the adipose tissue through mitochondrial ferritin (FtMT) overexpression in mice also prompts adipocytes to release smaller EVs such as exosomes [54]. The small EVs (sEV) are enriched with selective mitochondrial proteins including voltage-dependent anion channel (VDAC), heat shock protein 60 (HSP60), and cytochrome c oxidase subunit 4l1 (COXIV). Functional assessments of isolated sEVs from FtMT mice reveal that they are sub-competent in oxygen consumption when supplied with ADP, accompanied by elevated levels of carbonyl protein adduct formation. This leads to the thinking that oxidatively stressed adipocytes release sEVs containing partially functional and oxidatively damaged mitochondria. The sEVs containing oxidized mitochondria are found to circulate to cardiac tissues, where they are taken up by cardiomyocytes, integrated into the cardiomyocyte mitochondrial network, and eventually degraded.
The release of selective mitochondrial proteins through adipocyte EVs suggests that this process may be facilitated by mitochondrial vesicles (MDVs). Cells have the ability to sort distinct mitochondrial proteins into either single- or double-membrane MDVs [55]. Single-membrane MDVs emerge from the mitochondrial outer membrane, carrying with them cargo proteins located on the outer mitochondrial membrane, such as translocase of the outer mitochondrial membrane 20 (TOMM20). On the other hand, double-membrane MDVs form through the simultaneous budding of both the mitochondrial inner and outer membranes, transporting inner mitochondrial membrane and matrix proteins, such as pyruvate dehydrogenase (PDH). Research has revealed that single-membrane MDVs form through membrane protrusion driven by the mitochondrial Rho 1/2 (MIRO1/2) complex, which is subsequently pinched off by dynamin-related protein 1 (DRP1) [56]. Two factors known to enhance mitochondrial ROS production, namely xanthine oxidase and the complex III inhibitor antimycin A, have been shown to increase MDV formation [3]. Indeed, in both cold-stressed BAT and adipocytes in FtMT-overexpressing mice, there is a simultaneous increase in mitochondrial ROS production and MDV formation [53, 54]. These findings support the notion that MDVs play a pivotal role in EV-mediated release of defective mitochondrial components by the adipose tissue.
Mito-release triggered by defects in intracellular mitochondrial quality controlNumerous intracellular pathways contribute to the maintenance of mitochondrial quality in mammalian cells. Two notable pathways involved in this process are mitophagy and the MDV pathway. These pathways play a crucial role in targeting defective mitochondrial components to the lysosomes for degradation, thereby ensuring the maintenance of mitochondrial activity.
Recent reports have indicated that when the lysosome-dependent elimination of mitochondria is hindered, cells tend to increase the production of EVs to remove mitochondria from the intracellular space. For instance, the formation of the protein-lipid conjugate Atg8-phosphatidylethanolamine (Atg8-PE) is essential for mitophagy. Therefore, in HeLa cells expressing Parkin but lacking Atg8-PE formation, inhibiting mitochondrial functions did not result in the clearance of damaged mitochondria through the mitophagy pathway. Instead, these cells eliminated damaged mitochondria by releasing them into the extracellular space [57]. Another example involves the expression of disease-promoting mutations in Optineurin (OPTN), a protein associated with mitophagy. Mutations in OPTN are known to cause impaired or aberrant mitophagy [58, 59]. In neurons expressing mutant OPTN, transmitophagy, a process involving the release of mitochondria, was activated [46]. Additionally, inhibiting lysosomal acidification in mouse embryonic fibroblasts resulted in an increased secretion of mitochondria within large extracellular vesicles [60]. Similarly, hearts from aged mice or individuals with Danon disease exhibited elevated levels of secreted EVs containing mitochondria [60]. Danon disease is caused by mutations in the lysosome-associated membrane protein 2 gene, which impairs autophagic degradation. Lastly, in neurons derived from Huntington's disease (HD) patients, damaged mitochondria were found to be eliminated through EV-mediated extracellular release [61]. Consistent with this finding, the levels of neuron-derived extracellular vesicles containing mitochondria were enhanced in biofluids of HD patients. This observation is linked to the compromised mitophagy pathway commonly observed in cellular models of HD [62].
Taken together, these findings suggest that mammalian cells activate the extracellular release of mitochondria as a compensatory mechanism when mitophagy is impaired. This highlights extracellular release as a mitochondrial quality control mechanism in mammalian cells.
Comments (0)