Cell lines and cell culture
HeLa cells and HEK293T cells were cultured in high-glucose DMEM supplemented with 5% fetal calf serum (FCS) and 1% penicillin/streptomycin (P/S) under standard cell culture conditions. Human endothelial colony forming cells (hECFCs) were cultured in EBM-2 medium supplemented with 10% FCS and 1% P/S together with hydrocortisone, hFGF-B, VEGF, R3-IGF-1, ascorbic acid, GA-1000 and heparin from the EGM-2 Bulletkit (CC-4176, Lonza).
hECFC isolation and CD31 + flow cytometry analysis
hECFCs were isolated from human lung tissue according to Alphonse et al. with some modifications [19]. Lung tissue was obtained from residual, tumor-free material obtained from lung resection with the approval of the Medical Ethical Committee of the Erasmus MC Rotterdam. Lung tissue was either immediately processed or stored in high-glucose DMEM supplemented with 1% P/S overnight (O.N.) at 4 °C. Chopped lung tissue was digested with 7–10 ml of digestion solution (0.1 U/ml collagenase type I and 0.8 U/mg dispase in DPBS supplemented with CaCl2 and MgCl2) per 0.5 g of wet lung weight, and enzymes were quenched with an equal volume of high-glucose DMEM supplemented with 10% FCS and 1% P/S. Approximately 25 µl of CD31-dynabeads was used for up to 1–5*109 cells, and 24 h after isolation, the medium was refreshed, followed by refreshing three times a week until colonies appeared and were ready for further isolation. ECFC colonies were trypsinized with trypsin–EDTA (TE), which was quenched using 10% FCS/PBS and replated for expansion. The cells were purified for another 1–3 rounds with CD31-dynabeads until the cell population was > 95% CD31 + as determined by flow cytometry analysis on a BD LSRFortessa™ Flow Cytometer using a CD31-PeCy7 antibody (1:200, Biolegend) and DAPI (1:10,000, Beckton and Dickinson). After obtaining a pure ECFC population, the cells were either frozen or expanded for further experiments.
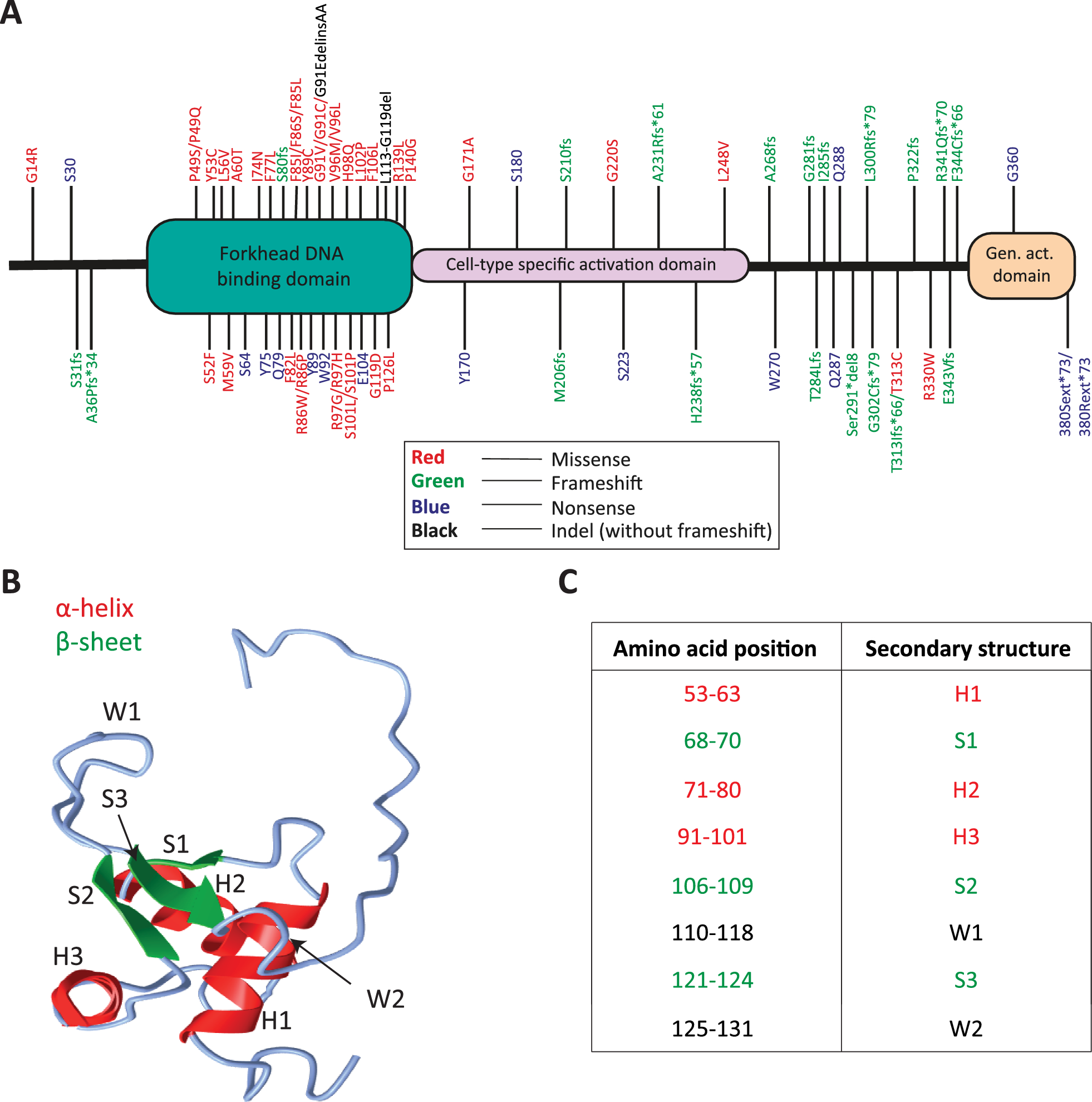
3D modelling of the FOXF1 DNA binding domain
Three-dimensional modelling of the FOXF1 DNA binding domain of FOXF1 was performed with EasyModeller 4.0 [20]. The protein sequence of the DNA binding domain of human FOXF1 (Supplementary Figure 1) was used as the query sequence, and FOXK2 (PDB ID: 1JXS), FOXO1 (PDB ID: 6qvw), FOXO3 (PDB ID: 2K86), and FOXO4 (PDB ID: 3L2C) were used as templates. Models were visualized using an iCn3D web-based 3D viewer [21].
Transfection
Cells were transfected using X-tremeGENE HP DNA Transfection Reagent (Roche) according to the manufacturer’s protocol. The ratio of DNA:X-tremeGENE HP DNA Transfection Reagent for HEK293T cells was 1:3 for immunoprecipitation and Phos-tag western blot experiments and 1:2 for other experiments. The ratio was was 1:2 for HeLa cells and 1:3 for HepG2 cells. Transfection complexes were added to cells in high-glucose DMEM supplemented with 1% FCS and 1% P/S for HeLa and HEK293T cells and cells were harvested 24 h after transfection. For HepG2 cells, transfection complexes were added in high-glucose DMEM supplemented 10% FCS 1% NEAA, 1% ultraglutamin and refreshed with complete medium after 24 h and cells were harvested 48 h after transfection.
Plasmids and cloningFOXF1 mutants
The wild-type FLAG-FOXF1 construct (CloneID OHu23845C) and a modified version of this plasmid lacking the stop codon were purchased from GenScript. The latter was used to construct a C-terminal V5-tagged FOXF1 plasmid by replacing the FLAG tag with a Kozak sequence and an ATG start codon and replacing the stop codon for a V5 tag. The C-terminal FLAG tag was made by replacing the V5-tag with a FLAG-tag at the C-terminus. DNA inserts were ordered as single-stranded oligonucleotides (IDT) and annealed to make double-stranded DNA inserts. Single-stranded oligonucleotide sequences are listed in pJ3M-MST1 (Addgene plasmid: #12203), pJ3MST1 K59R (Addgene plasmid: #12204), pJ3M-MST2 (Addgene plasmid: #12205) and pJ3M-MST2 K56R (Addgene plasmid: #12206) were gifts from Jonathan Chernoff, MD, PhD [22]. MST1 and MST1 K59R were cloned from the pJ3M plasmid into the pcDNA3.1 + plasmid by restriction with HindIII and EcoRI. To clone MST2 and MST2 K56R into pcDNA3.1 + , pJ3M plasmids were digested with SalI and EcoRI, and the pcDNA3.1 + plasmid was digested with HindIII and EcoRI. Blunt ends were created with Klenow, and MST1 and MST2 K56R inserts were ligated into the pcDNA3.1 + vector.
Table 1.
Table 1 Oligonucleotide sequences for cloningThe L56V mutant was incorporated into either wild-type FLAG-FOXF1 of FOXF1-FLAG using the QuickChange Site-Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instructions with the following modifications to the PCR mix and PCR cycling parameters: for the PCR, 500 ng of dsDNA template was used instead of 5–50 ng, and 10% DMSO was added to the PCR mixture. For PCR cycling, the first step consisted of 1 cycle at 95 °C for 3 min instead of 1 cycle at 95 °C for 30 s. The primers used are listed in Table 2.
Table 2 PCR primers used for constructing the L56V mutationAll other mutants were constructed using GenParts Elite DNA fragments. For each mutation, a DNA fragment was ordered with the BlpI and BsrGI restriction sites, and the digested fragments were exchanged with the wild-type fragment from the FLAG-FOXF1 or FOXF1-FLAG plasmid. The sequences of the DNA fragments are included in Table 3.
Table 3 Elite double-stranded DNA fragments used for constructing FOXF1 missense mutants in the F85-S101 regionpJ3M-MST1 (Addgene plasmid: #12203), pJ3MST1 K59R (Addgene plasmid: #12204), pJ3M-MST2 (Addgene plasmid: #12205) and pJ3M-MST2 K56R (Addgene plasmid: #12206) were gifts from Jonathan Chernoff, MD, PhD [22]. MST1 and MST1 K59R were cloned from the pJ3M plasmid into the pcDNA3.1 + plasmid by restriction with HindIII and EcoRI. To clone MST2 and MST2 K56R into pcDNA3.1 + , pJ3M plasmids were digested with SalI and EcoRI, and the pcDNA3.1 + plasmid was digested with HindIII and EcoRI. Blunt ends were created with Klenow, and MST1 and MST2 K56R inserts were ligated into the pcDNA3.1 + vector.
Constructs for luciferase assays were generated by cloning 5 repeats of the RTAAACA binding motifs or scrambled motifs separated by 10 nucleotides into the pGL4.10 [luc2] vector (Promega; E6651). DNA inserts were ordered as single-stranded oligonucleotides (IDTs) and annealed to make double-stranded DNA inserts, which are listed in Table 4. To each primer, a restriction site was added, 5’ for AccI and 3’ for XhoI, to clone the binding motif into the pGL4.10 [luc2] vector.
Table 4 sequences of Primers used for cloning the FOXF1 binding motifs into the pGL4.10[luc2] vectorImmunofluorescence staining
Transfected HeLa cells on coverslips were fixed 48 h post-transfection with 4% paraformaldehyde for 10 min at room temperature (RT), permeabilized with 0.1% Triton-X/PBS for 10 min at RT and incubated for 20 min in blocking buffer (3% BSA in 0.05% Triton-X/PBS). Primary antibodies against FLAG (1:250, F7425 Sigma) were diluted in blocking buffer and incubated O.N. at 4 °C. The cells were subsequently washed and incubated for 1 h with fluorophore-conjugated secondary antibodies at RT. Subsequently, the cells were incubated with DAPI (1:2000, 564907; Becton and Dickinson) for 10 min at RT, and the coverslips were mounted using Mowiol 4–88 (81381 Sigma). The samples were imaged using a Leica TCS SP5 confocal laser scanning microscope and analysed using Fiji software.
Protein extraction, SDS‒PAGE and phos-tag western blotting
For SDS‒PAGE, protein extracts of HEK293T cells were made as previously described [23], except for phosphorylation experiments, where RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% Na-DOC, 0.1% SDS, 50 mM Tris pH 7.5) with complete EDTA-free protease inhibitor (CEF) (Roche) and PhosStop (Sigma) were used as protein extraction buffers for SDS‒PAGE. Proteins were separated by gel electrophoresis using a 4–12% ExpressPlus™ PAGE gel (GenScript) and transferred onto a PVDF membrane (Immobilon).
Phos-tag western blotting was performed according to the phos-tag product (AAL-107 M, Wako) manual protocol of Wako using the MnCl2 method with some modifications. Briefly, standard protein extracts of HEK293T cells were made in RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% Na-DOC, 0.1% SDS, 50 mM Tris, pH 7.5) supplemented with complete EDTA-free protease inhibitor (CEF) (Roche) and PhosStop (Sigma). For phosphatase treatment, PhosStop was excluded from the protein extraction buffer, and the protein extract was incubated with rSAP (NEB) for 1 h at 37 °C. Proteins were separated by gel electrophoresis on a 10% running gel with 100 µM phos-tag using Tris–glycine buffer and transferred onto a PVDF membrane (Immobilon).
PVDF membranes were blocked using 3% BSA in 0.05% Tween in TBS and labelled with primary antibodies against FOXF1 (R&D, AF4798), MYC-tag (Abcam, ab9106), FLAG-tag (Sigma, F7425), HA-tag (Santa Cruz, sc-805), MST1 (Abcam, ab51134), MST2 (Abcam, ab52641) or cofilin (Abcam, ab42824) followed by HRP-conjugated secondary antibody labelling. Blots were developed using Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific) and imaged on an Amersham Imager 600 (GE Healthcare).
Co-immunoprecipitation experiments
Whole-cell lysates were made from transfected cells as described previously [23]. Co-immunoprecipitation experiments with WT expression constructs were performed with protein G agarose beads as previously described [23] or with Dynabeads™ Protein G Immunoprecipitation Kit (Invitrogen, 10007d) according to the manufacturer’s instructions for experiments with WT and kinase-dead expression constructs. Immunoprecipitation was performed with antibodies against MYC-tag (Roche, 1668149), FLAG-tag (Sigma, F1804) and HA-tag (Santa Cruz, sc-7392).
Chromatin immunoprecipitation (ChIP) and motif analysis
A total of 20 × 106 hECFCs were crosslinked in 1% formaldehyde, which was quenched with 0.125 M glycine. The cells were pelleted by centrifugation and snap-frozen for storage or sonicated. The cells were lysed in SDS lysis buffer (50 mM Tris, pH 8; 10 mM EDTA; 1% SDS; 1 × CEF), and the chromatin was sonicated for 15 cycles (30 s on, 30 s off per cycle) using a Bioruptor Pico (Diagenode). Chromatin was diluted with ChIP dilution buffer (167 mM NaCl, 16.7 mM Tris–HCl (pH 8), 1.1% Triton-X, 1.2 mM EDTA, 0.01% SDS with 1 × CEF). BSA-blocked protein G agarose beads (Millipore) were incubated O.N. at 4 °C with antibodies (FOXF1 AF4798 R&D or goat IgG R&D) and added to diluted chromatin for 3 h at 4 °C under gentle rotation. The beads were subsequently washed with low-salt immune buffer (150 mM NaCl, 20 mM Tris–HCl (pH 8), 2 mM EDTA, 1% Triton X, 0.1% SDS), high-salt immune buffer (500 mM NaCl, 20 mM Tris–HCl (pH 8), 2 mM EDTA, 1% Triton X, 0.1% SDS), LiCl immune complex buffer (250 mM LiCl, 10 mM Tris–HCl (pH 8), 1 mM EDTA, 0.5% NP-40, 0.5% Na-DOC) and TE buffer (10 mM Tris–HCl (pH 8), 1 mM EDTA). Bound chromatin was eluted with elution buffer (300 mM NaCl, 10 mM Tris–HCl (pH 8), 5 mM EDTA, 0.5% SDS) supplemented with 0.29 mg/ml proteinase K and incubated O.N. at 65 °C to decrosslink the chromatin. DNA was purified using a Qiagen MinElute column, and DNA libraries were prepared using the ThruPLEX DNA sample preparation protocol from Takara Bio and sequenced on an Illumina HiSeq2500 sequencer. Adapter sequences were removed from the sequence reads. These were aligned were aligned to the human GRCh38 reference genome using the HISAT2 aligner [24]. After alignment, duplicate reads were filtered and peaks were called with the MACS2 peak caller [25]. Sequence coverage over the genome was determined. For motif analysis, only peak sequences in promoter regions defined as 1 kb from transcription start sites were used, and motifs were identified with MEME-ChIP [26, 27].
CUT&TAG
CUT&TAG experiments were performed with hECFCs according to a previously published protocol from the Henikoff laboratory, version 3 [28]. In brief, hECFCs were cultured until confluency, and a total of 11 × 106 cells were harvested in 10% FCS/PBS and counted. Cells were isolated using Concanavalin A-coated (BP531-3 ml, Sanbio) beads. Primary antibodies against FOXF1 (AF4798, R&D) or the IgG control (AB-108-C, R&D) were incubated overnight at 4 °C, and the membranes were incubated with secondary antibodies for 1 h at room temperature. pA-Tn5 adapter complexes (C01070001, Diagenode) were bound at room temperature for 1 h, and tagmentation was performed for 1 h at 37 °C. DNA was extracted using the chloroform extraction method, and PCR was performed using NEBNext HiFi 2 × PCR master mix (M0541S) and indexed i5 and indexed i7 primers (Table 5). DNA was purified after PCR using SPRI paramagnetic beads (Agentcourt AMPure XP, A63880) and checked using a High DNA Sensitivity DNA assay on an Agilent Bioanalyzer 2100. Each sample was analyzed in duplicate, and DNA libraries and subsequent filtering and peak calling were performed as described above.
Table 5 CUT&TAG PCR primersElectrophoretic mobility shift assay
EMSA assays were performed using the LightShift™ Chemiluminescent EMSA Kit (20,148 Thermo Scientific) according to the manufacturer’s instructions. Protein extracts were made as described for western blotting in Carin buffer. Briefly, all the samples were subjected to binding reactions and incubated for 20 min at RT. For binding reactions, biotin-labelled or nonlabelled DNA probes were used, and the sequences are listed in Table 6. Probes were ordered as single-stranded oligonucleotides and annealed to produce double-stranded DNA probes. After the binding reaction, the samples were loaded onto a native polyacrylamide gel with a ratio of 29:1 of acrylamide:bisacrylamide. First, 1 ml of 10 × Tris/borate/EDTA (TBE) buffer (900 mM Tris, 900 mM boric acid, 20 mM EDTA, pH 8.3), 2 ml of 40% acrylamide, 1.35 ml of N,N’-methylenebisacrylamide 2%, 200 µl of 10% ammonium persulfate and 20 µl of N,N,N,N-tetramethylenediamine were added to 15.43 ml of ultrapure water, and the mixture was run for 45 min at 100 V until ¾ of the gel. DNA was transferred to a Hybond XL nylon membrane (GE Healthcare) for 30 min at 380 mA and crosslinked to the membrane using a transilluminator equipped with 312 nm bulbs for 10 min. The membrane was blocked using blocking buffer included in the supplier’s kit and incubated with streptavidin–horseradish peroxidase conjugate. After the membrane was washed, the substrate equilibration and substrate working solution from the kit were added, and the membrane was imaged on an Amersham Imager 600 (GE Healthcare).
Table 6 Primer sequences for the EMSA DNA probesLuciferase assays
A luciferase assay was performed as previously described [29] with several modifications. HeLa cells were transiently transfected with Lipofectamine 3000 (Thermo Fisher, L3000001). Each transfection consisted of 50 ng of pcDNA3 expression plasmid, 50 ng of pGL4.10[luc2] reporter plasmid and 10 ng of TK-Renilla plasmid (Promega; E2241). Luciferase activity was measured 24 h after transfection using the Dual-Luciferase Reporter Assay System (Promega, E1910), and luminescence was measured with a VICTOR X4 plate reader. Each sample was normalized for transfection efficiency. An increase or decrease in luciferase activity was determined by normalizing luciferase activity to that of the scrambled motif control.




Comments (0)