
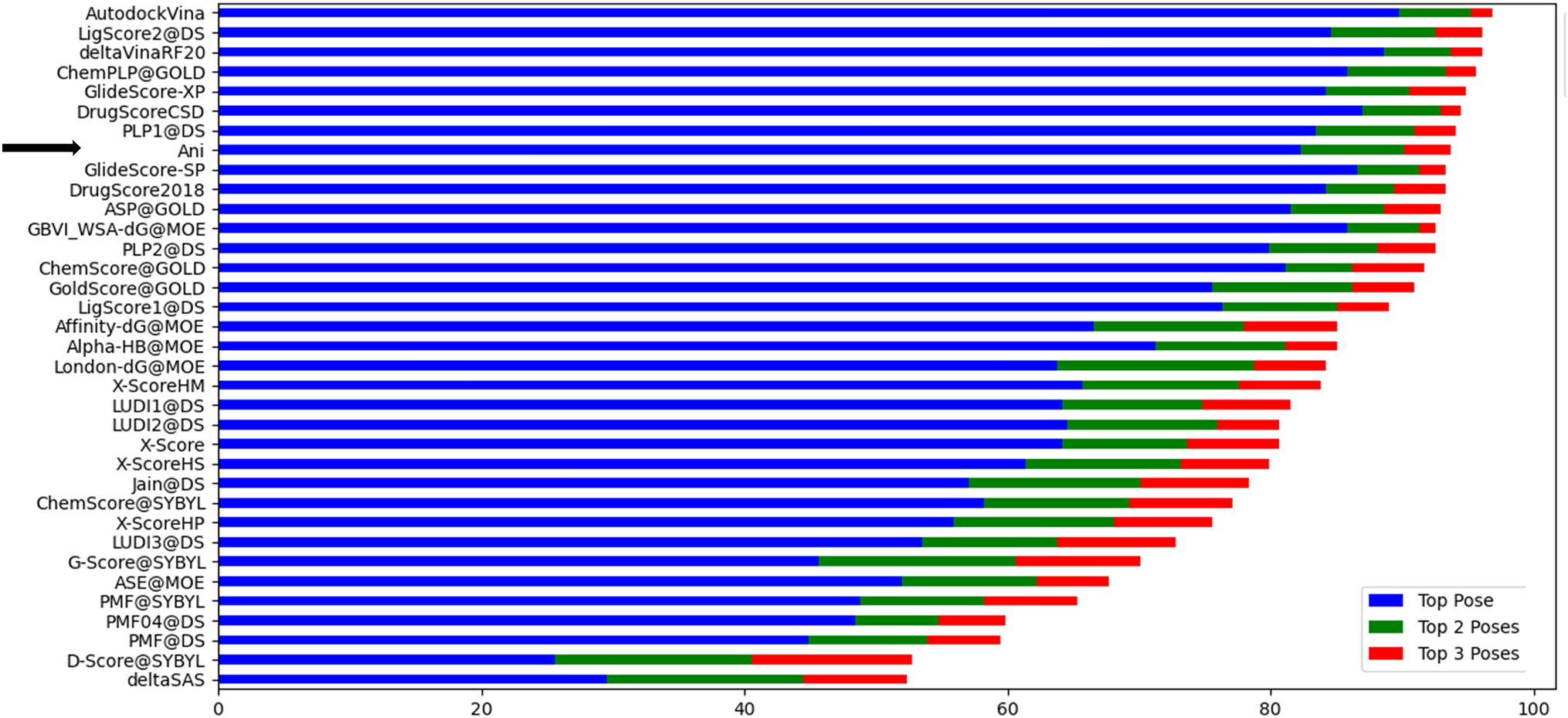
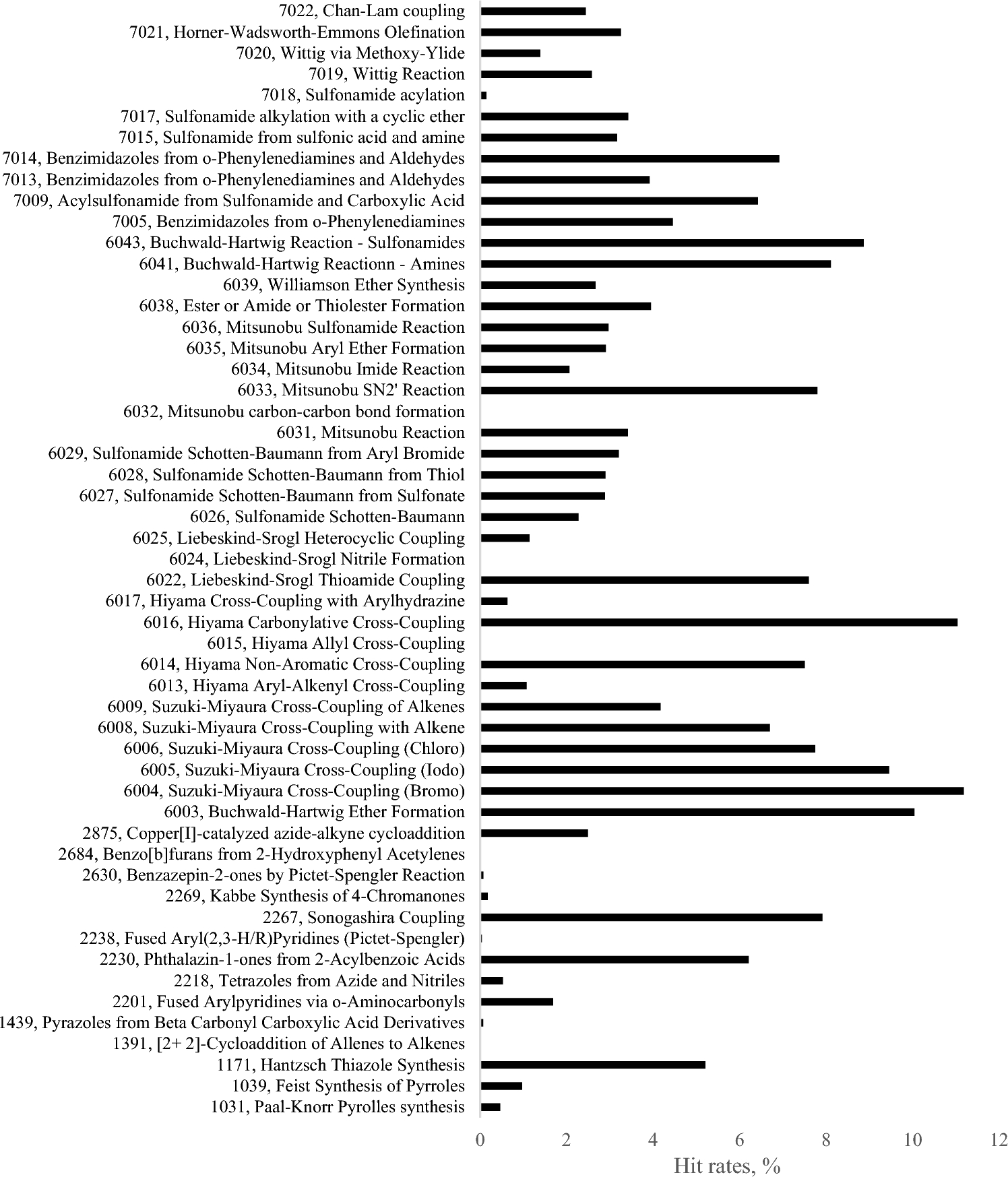
Remember me

The PIE between ligand and receptor fragments, EINT, is widely used in FMO LR study. In a previous work on Au(I)-biscarbene/Gq complexes [20], we experienced that the use of the most applied FMO computational settings, i.e., the FMO2 RI-MP2/6-31G* level of theory, leads to huge and unrealistically negative EINT and ΔEFMO2 values when compared with experimental binding energy of − 10.4 kcal/mol (Kd = 0.03 µM) [26, 49]. There, we showed that this issue is related to the overestimation of the CT energy occurring when metal atoms are included in the target structure. We also found that, compared with local model, the partial screening method was necessary to properly account for the solvent screening effect and to improve the accuracy of EINT of 1 and 2. Nevertheless, its adoption did not mitigate the overestimation of CT energy. In this section, to overcome this problem we analyse the impact of several factors, namely the level of theory, the basis set, the type of n-body approach and the use of screened point charges for all atoms in ESP-PTC calculation (ESP-SPTC), on EINT values. The results are summarized in Table 1 and S1.
Table 1 EINT values computed for Gq-1 and Gq-2 complexes at FMO2 RI-MP2/6-31G//sc, FMO2 RI-MP2/6-31G*//sc, FMO2 RI-MP2/6-311G//sc and FMO2 RI-MP2/6-311G//sc levels of theory. All values are in kcal/molWe preliminary evaluated the effect of the electron correlation comparing the EINT values computed at FMO2 HF/6-31G* level of theory (Table S1) with those computed using the RI-MP2 method (FMO2 RI-MP2/6-31G*). Results show a relevant impact of correlation (Fig. S3A) which is important to correctly describe many-electrons system and to compute the dispersion interactions (Edisp). Therefore, we evaluated the effect of the other computational features on EINT only at RI-MP2 level of theory.
The polarization (d) functions are important to increase the mathematical flexibility of the wave functions and to provide a better description of several chemical aspects (e.g., dipole moments, anions). To assess their impact, we compared the EINT values computed with 6-31G and 6-31G* at RI-MP2 level of theory. As shown in Fig. 2a the inclusion of d functions determines a slight increase of the binding strength (more negative EINT) with no significant change in their magnitude.
The same effect, though to a larger extent, was found by expanding the basis set from 6-31G to 6-311G (Fig. 2b): EINT terms become significantly more negative, even assuming an unrealistic value for Gq(I)-2 of −752.9 kcal/mol. The same conclusions can be obtained by comparing EINT at 6-31G* and 6-311G at RI-MP2 level of theory (Fig. 2c).
Thus, the application of polarization d functions or the triple-ζ basis set (6-311G) does not sensibly improve the accuracy of EINT which remains significantly more negative than experimental binding energy.
As mentioned in the Theoretical background section, the ESP can affect the charge transfer between i and j in dimer ij. In our previous work [20] the ESP was computed considering the point charge approximation (ESP-PTC) only between fragments exceeding the VdW factor 4.0 (RESPPC = 4.0) while the two electrons integral contribution was included for ESP between fragments below this value (ESP2). Notably, the ESP2 generally provides a more accurate description of ESP and therefore is generally the most applied approach [26].
We use here an alternative strategy in which the ESP-PTC is described by screened point charges for all atoms adopting the dumping function (ESP-SPTC), Eq. 8, setting a = b = 1, as suggested by the GAMESS-US manual. We also tried a = 1 and b = 2 but observed a negligible change in EFMO and EINT values as already reported by Fedorov et al. [26].
As shown in Fig. 2d, EINT computed at FMO2 RI-MP2/6-31G*//sc are significantly smaller than the corresponding data found with FMO2 RI-MP2/6-31G* and the same results were found with 6-311G basis set (Fig. 2d). For instance, EINT for Gq(I)-1 passes from − 326.6 to -36.5 kcal/mol, a more reasonable value closer to experimental reference data (− 10.4 kcal/mol). The relevant role in reducing the EINT magnitude exerted by the ESP-SPTC is also observed for results obtained with the 6-31G basis set (Fig. S3B).
The use of the triple-ζ basis set leads to a slight increment of EINT (Fig. 2e) but the value for Gq(I)-2 remains positive as found at FMO2 RI-MP2/6-31G*//sc level of theory.
The investigation of the complex between the Trp-cage protein bound with deprotonated p-phenolic acid highlights that the three-body approach corrects the overestimation of the CT energy in FMO2 and leads in general to more accurate PIE values [28]. However, we found that the application of the FMO3 approach alone was not enough to obtain more accurate EINT values, confirming the requirement of the screened point charges in the ESP-PTC calculation, as shown in Fig. 2f.
Thus, while the binding affinity of ligand 1 is well described by EINT computed at FMO2 RI-MP2/6-31G*//sc level of theory, for ligand 2 the best results have been obtained by employing the FMO3 approach with the 6-311G basis set (Fig. 2g), which determines more negative values of EINT compared to those obtained with the 6-31G* basis set.
Fig. 2
Comparison between EINT values for ligands 1 and 2 at Gq(I), Gq(II) and Gq(III) binding sites, computed using different levels of theory: a FMO2 RI-MP2/6-31G vs. FMO2 RI-MP2/6-31G*; b FMO2 RI-MP2/6-31G vs. FMO2 RI-MP2/6-311G; c FMO2 RI-MP2/6-31G* vs. FMO2 RI-MP2/6-311G; d FMO2 RI-MP2/6-31G* vs. FMO2 RI-MP2/6-31G*//sc; e FMO2 RI-MP2/6-311G vs. FMO2 RI-MP2/6-311G//sc; f FMO3 RI-MP2/6-31G* vs. FMO3 RI-MP2/6-31G*//sc; g FMO3 RI-MP2/6-31G*//sc vs. FMO3 RI-MP2/6-311G//sc and h FMO2 RI-MP2/6-311G//sc vs. FMO3 RI-MP2/6-311G//sc. All energy values are reported in kcal/mol
As shown in Fig. 2h, the EINT calculated at FMO3 RI-MP2/6-311G//sc is characterized by the same trend found with FMO2 RI-MP2/6-311G//sc but with a significant improvement only for Gq(I)-2. Indeed, the corresponding EINT is now negative (−1.0 kcal/mol), suggesting that the description of the interaction of ligand 2 at the position I requires a higher-level computational approach than FMO2 (Fig. 2g). Notably, the use of the FMO3 approach with low accuracy would require large basis set as 6-31G**, 6-311G*, 6-311G** (or larger) to produce results more accurate than those obtained with the FMO2 method. However, these basis sets significantly increase the computing time, making their application to routine CADD studies very difficult. Therefore, to assess the impact of the ESP-SPTC combined with such large basis sets using the FMO3 (low accuracy) we computed EINT of ligand 1, with and without the screened point charges, considering a reduced model of the Gq receptor represented only by G nucleobases and K+ ions (Fig. S4). Results reported in Table 2 show that even with larger basis the FMO3 (low accuracy) alone does not provide accurate EINT values. On the contrary, the application of ESP-SPTC allows obtaining more reliable results, closer to experimental binding energy.
Table 2 EINT values computed for the reduced model of Gq(I)-1 (Fig. S4) using the FMO3 (low accuracy) method at RI-MP2/6-31G**, RI-MP2/6-311G*, RI-MP2/6-311G**, RI-MP2/6-31G**//sc, RI-MP2/6-311G*//sc and RI-MP2/6-311G**//sc levels of theory. All values are in kcal/molThe EDA of EINT provides a crucial insight on the nature of bonding and allows monitoring how the contribution of CT energy varies as a function of the basis set magnitude and upon the use of the ESP-SPTC. In Fig. 3 and in Table S2 we reported the EDA for ligand 1 at the three binding poses while the corresponding values for ligand 2 are reported in Fig. S5 and Table S3.
Fig. 3
Bar diagram of total PIEDA for Gq-1, considering the three different binding regions I, II and III, computed at the FMO2 RI-MP2/6-31G*, FMO2 RI-MP2/6-31G*//sc, FMO2 RI-MP2/6-311G//sc and FMO3 RI-MP2/6-311G//sc levels of theory. Ees, Eex, Ect, Edisp and Esol are the electrostatic, exchange repulsion, charge transfer, dispersion and solvation energies, respectively
Figure 3a shows the impact of the ESP-SPTC on the EDA terms at the FMO2 RI-MP2/6-31G* level of theory. Ees, Eex and Ect are significantly influenced by the screened point charges that markedly decrease the absolute value of these energy contributions. Ect is the most affected term passing from −219.3, −127.1 and −109.0 to −17.0, −20.5 and 19.7 kcal/mol for Gq(I)-1, Gq(II)-1 and Gq(III)-1, respectively. This evidence highlights the crucial contribution of the ESP-SPTC to provide correct estimates of the Ect term.
The use of the 6-311G basis set and of FMO3 approach leads in general to more negative EINT values, with relative weights of Ees, Eex, Ect, Edisp and Esolv contributions that resemble those obtained at FMO2 RI-MP2 6-31G*//sc (Fig. 3b).
A similar situation is found for the EDA of EINT performed for ligand 2 (Fig. S5 and Table S3). In this case, as mentioned before, the FMO3 treatment results to be crucial to correctly estimate the attractive contribution of Ect (−22.8 kcal/mol) in Gq(I)-2 complex, even with the 6-311G basis set, for which we found a positive value of +14.2 kcal/mol.
According to Eq. 15, EINT can be decomposed into three terms,\(\sum _^(_^}- _^}-_^})\), \(\sum _^_^\) and \(\sum _^_^\) where the latter term is the embedded charge transfer energy (Tr(ΔDij*Vij)) between ligand and all Gq fragments. As shown in Fig. 4 and reported in Table S4, Eemb is the energy term that changes most by applying the screened point charges for all atoms, from large negative (attractive) to small positive values. Indeed, passing from FMO2 RI-MP2/6-31G* to FMO2 RI-MP2/6-31G*//sc level of theory this term changes from −222.4, −94.9 and −98.8 to 2.2, 2.8 and 1.4 kcal/mol for Gq(I)-1, Gq(II)-1 and Gq(III)-1, respectively, confirming that the ESP-SPTC is crucial to fix the embedded CT overestimation of pair interactions involving metals-containing fragments.
Fig. 4
Bar diagram of EINT decomposed according to Eq. 15 for Gq-1, considering the three different binding regions I, II and III, at the FMO2 RI-MP2/6-31G* and FMO2 RI-MP2/6-31G*//sc levels of theory. Esol, Eemb and (ELi - EL - Ei) are the solvation energies, the embedding energy (that is the sum of ligand-receptor embedded charge transfer energies) and the energy difference between the internal solute energy respects with Li complex, respectively
Ligand 1 with the nearest fragments containing K+, i.e., fragments 7 and 19 for Gq(I) and Gq(II)/Gq(III), respectively, shows the most significant Ect decrease, with a consequent improvement of the EINT accuracy, as shown by diagrams reported in Fig. 5a, b and c. The PIE of fragment 7-ligand 1 calculated with the 6-31G* basis set is −137.9 kcal/mol which becomes 4.0, 2.5 and 2.6 kcal/mol by adopting the screened point charges for all atoms at FMO2 RI-MP2/6-31G*, FMO2 RI-MP2/6-311G and FMO3 RI-MP2/6-311G levels of theory (Fig. 5d, e and f and Table S5), respectively. As reported in our previous work (Table S2 of ref. 19), the Ees contributions to the PIE between the two positively charged fragments, i.e., 1 and fragment 7/fragment 19 in Gq(I) computed at FMO2 RI-MP2 6-31G*, are negative values, leading to some inconsistency. On the contrary, the inclusion of the screened point charges yields positive Ees values, correctly indicating a repulsive electrostatic interaction between two fragments with the same charge. All other energy terms, except Esolv, become negligible with values close to zero.
As reported in Table S6, the same favourable effects of ESP-SPTC on Ees and Ect terms were observed in PIEs between 1 and (G22∙G10∙K102) fragment computed for reduced Gq(I)-1 complex with FMO3 (low accuracy) using large basis set (6-31G**, 6-311G* and 6-311G**).
Notably, the impact of ESP-SPTC can be also appreciated by considering amount of CT, QCT, from ligand to the nearest fragment containing K+ ion (Table S5-S7). Indeed, for instance, for Gq(I)-1 the QCT are −0.2095 and −0.0126 at FMO2 RI-MP2/6-31G* and FMO2 RI-MP2/6-31G*//sc, respectively, suggesting that ESP-SPTC might effectively reduce the overestimation of CT. The analysis of other QCT values confirms this trend.
Comparable results are also found for ligand 2 (Fig. S6, Table S3 and S7), although a positive value of EINT is obtained for Gq(I)-2 at FMO2 RI-MP2/6-31G*//sc.
By applying the ESP-SPTC, the profile of PIEs chart is significantly simplified where the most relevant interactions are limited to those between the ligand and the underlying nucleobases (Fig. 5), which are G5–G11, G15–G21 and G3–G9 at Gq(I), Gq(II) and Gq(III) binding sites, respectively.
As shown in Fig. 3, the main contribution to EINT is represented by electrostatic energy, with a lower contribution of Edisp and Ect. These two terms are significative only in the pair interactions between ligands and the underlying nucleobases DG5, DG11 DG15, DG21, DG9 and DG3 (Table S8) which are the closest to the ligands. The EDA of the interactions between the ligand and these nucleobase pairs (Tables S8 and S9) confirms the asymmetric interaction of ligands 1 and 2 as reported in the previous work [20], supporting the occurrence of π-cation interaction hypothesized for 1.
Fig. 5
\(_^\) values for the interaction between Gq fragments and ligand 1 in the binding sites I (A and D), II (B and E) and III (C and F), computed at the FMO2 RI-MP2/6-31G*, FMO2 RI-MP2/6-31G*//sc, FMO2 RI-MP2/6-311G//sc and FMO3 RI-MP2/6-311G//sc levels of theory and reported by using red, green, blue and orange lines, respectively
The average PIEs between ligands and the underlying guanines pairs are in good agreement with the average EINT values (Table S10) suggesting that this specific interaction energy can be used to quickly assess the binding affinity of a set of Gq binders, especially if the FMO method is combined with either molecular dynamics or other multi-conformational approaches where the execution of many FMO calculations is needed.
The average EINT values defined for Gq-1 and Gq-2 complexes are −50.2 and −30.2 kcal/mol, respectively, at FMO3 RI-MP2/6-311G//sc level of theory and −45.7 and − 12.9 kcal/mol at FMO2 RI-MP2/6-311G//sc, well reproducing the trend of the experimental binding efficiency of two Au(I)-binders investigated in this work (Table 2). For ligand 1, the average EINT value closest to experimental binding energy (−10.4 kcal mol−1) was instead computed at FMO2 RI-MP2/6-31G*//sc level of theory (−34.9 kcal/mol).
The present results indicate that the use of the EPS-SPTC should be considered to correctly describe the CT energy in a peculiar system as the Au(I)-biscarbene/Gq complexes. This evidence agrees with the methodological study of water clusters performed by Fedorov et al. [26] where a worse performance of ESP2 compared to ESP-PTC was found, the more so with the increasing of basis set size. The same effect is shown here when passing from double-ζ to triple-ζ basis sets. ESP2 can promote the convergence to an unphysical electronic state with consequent abnormal CT between the two fragments [20, 26]. Finally, again in agreement with the results of Fedorov et al. [26], the relevant improvement in the accuracy produced by the charges’ screening can be explained with their capability to significantly reduce the polarization of the electronic state at short distance [26]. However, for common applications the default EPS-PTC/ESP2 approach should always be considered as the reference method.
As a final remark, it is worth noting that Gq/Au(I)-biscarbene complexes represent a challenging system from the computational point of view where two K+ ions are in a channel surrounded by negatively charged sugar-phosphate skeleton and the positively charged Au(I) ligands are located perpendicular to the vertical axis connecting the two K+ ions. This peculiar geometrical configuration, where the distance between ligands and nearest K+ ions is 5–6 Å (Fig. S7), might promote charge transfer, making the correct evaluation of the related energy difficult.
FMO binding energy, ΔEFMOThe ΔEFMO, conversely from EINT, considers the polarization-destabilization and desolvation energies of both ligand and receptor passing from free to bound state, providing in principle a better description of the binding process.
According to Eq. 1, ΔEFMO is strictly related to EINT. Therefore, we computed ΔEFMO by using the best computational settings found for EINT such as the FMO2 RI-MP2/6-31G* and FMO3 RI-MP2/6-311G levels of theory combined with the ESP-SPTC.
As shown in Table S11, the use of screened charges for all atoms has a huge effect on ΔEFMO values calculated at FMO2 RI-MP2/6-31G* and FMO3 RI-MP2/6-311G levels of theory, significantly reducing their magnitude as find for EINT. Both FMO2 and FMO3 binding energies computed with ESP-SPTC are positive values in the most cases, hampering a direct comparison with experimental binding data. This result might also suggest that the adoption of the ESP-SPTC for all atoms might lead to a possible slight underestimation of interaction energies. The employ of a large basis set, facilitated by the auxiliary polarization, FMO/AP [50], might improve the accuracy of ΔEFMO values computed using the ESP-SPTC.
It should be remarked that ΔEFMO does not include entropy that could give a crucial contribution to the binding, especially when hydration entropy is considered. In the formation of the LR complex, both isolated ligand and receptor release the hydration water molecules with subsequent increase of entropy. The hydration entropy was indeed found to be crucial to determine negative energies for the binding of small peptides to RNA quadruplex [51]. However, considering that ligands 1 and 2 are small molecules compared with a peptide, the real contribution to the hydration entropy for biscarbene-Au(I)/Gq binding process should be carefully evaluated and could not be enough to compensate the positive ΔEFMO value.
In the present case, where no entropic contributions were considered, the EINT values computed with the ESP-SPTC show a better agreement with experimental evidence than ΔEFMO values, even at FMO2 level, representing therefore the best term to describe the binding affinity of metal-containing ligands to DNA-Gq. The description of systems like biscarbene-Au(I)/Gq might also benefit from the application of partition analysis method based on charge transfer state with fractional charges proposed for the DFTB [52].
In summary, the computational protocol to assess the binding efficiency of metal-binders to DNAGq by adopting the FMO method should include (i) the partial screening method to correctly simulate the solvent screening effect and should consider the application of (ii) the ESP-SPTC for all atoms when questionable PIE values are obtained using the default ESP treatment. Combined with these features, the FMO2 RI-MP2/6-31G* level of theory provides satisfactory results for EINT while the adoption of larger basis set might improve the accuracy when it is coupled with FMO3 approach. However, we suggest applying the three-body approach only in critical cases where the FMO2 method does not ensure enough accuracy, like structures containing many charged fragments close to each other.
It is finally worth noting that the accuracy in the prediction of a set of ligands binding affinity based on EINT can be significantly improved by its combination with additional energy terms, e.g., entropic and hydrophobic terms. A recent study, where EINT was linearly combined with clogP, demonstrated that a similar approach can be effectively used to predict with great accuracy (R2 = 0.9) the binding efficiency of ligand-receptor systems not containing metal atoms [53]. Our future work will be devoted to developing a scoring function based on our FMO/GRID approach [7, 10] specifically designed to predict the binding energy of LR complexes containing metals.
Comments (0)