
Remember me


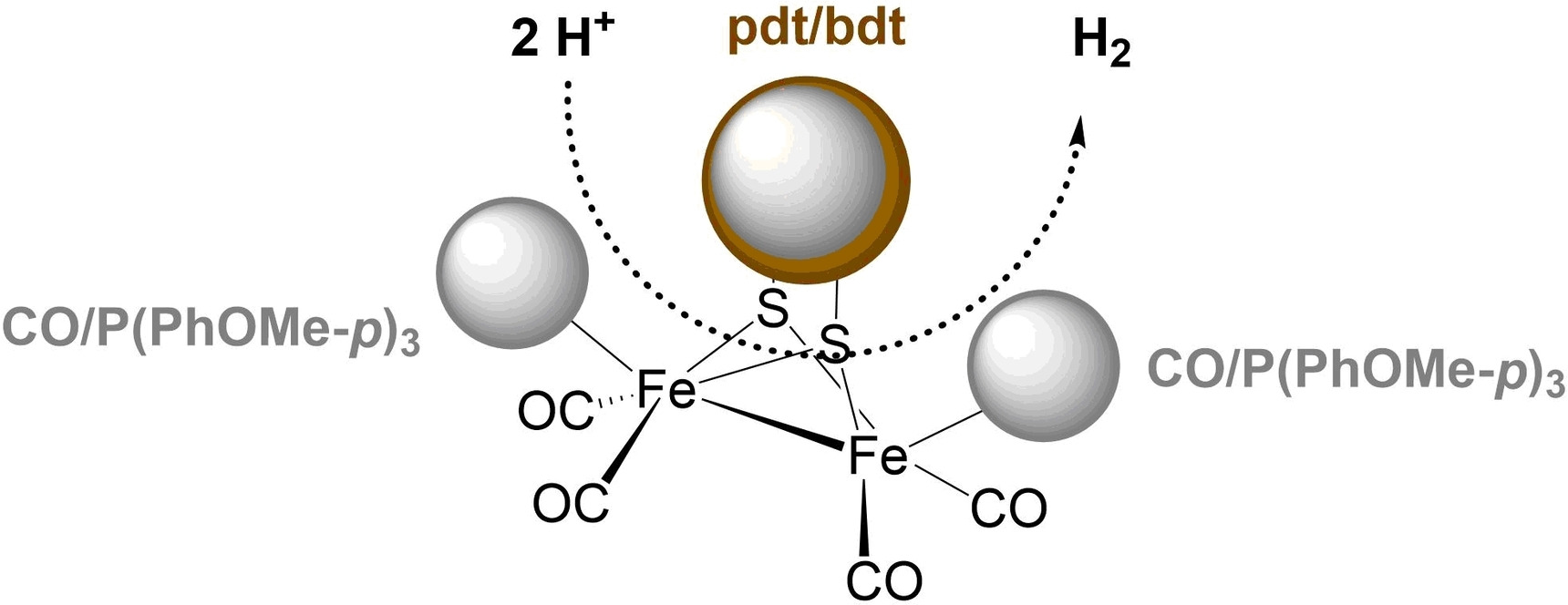
Cyclic voltammograms (CVs) for complexes 1–4 were measured in acetonitrile under an argon atmosphere (see Table 1). The CVs for 1 display two irreversible reduction events at Epc=−1.87 and −2.37 V and two irreversible oxidations at Epa=0.35 and 0.70 V (Figure S9, see Supporting Information). On the other hand, only one irreversible reduction at −1.65 V is observed for complex 3 (Figure S9). The reductions for complexes 1 and 3 occur at more negative potentials than for the all-carbonyl complex due to the substitution of one CO ligand with the electron-donating phosphine ligand.42, 44, 60 Also, the reduction of 3 occurs at a less negative potential and its oxidation at a more positive potential than 1 due to the aromatic bdt ligand in 3. The electrochemical data are consistent with the results of similar model complexes reported in the literature.58b
Table 1. Experimental and calculated electrochemical data for complexes 1–4.Complex
Epc/V
Epa/V
Ecat/V[c]
Calculated Epc/V[d]
1
−1.87
0.35
−1.79
−1.81
−2.37
0.70
−1.96
−2.46
2
−2.10
0.27
−1.65
−2.37
−2.51
–
−1.99
−2.60
3
−1.65
0.49
−1.57
−1.60
–
–
−1.91
−1.77
4
−2.02
0.39
−1.43
−1.90
–
0.67
−1.71
−2.09
[Fe2(CO)6(μ-pdt)]
−1.74[a]
–
–
−1.48
−2.35[a]
–
–
−2.15
[Fe2(CO)6(μ-bdt)]
−1.31[b]
–
–
−1.32
[a] In dichloromethane; [b] ; [c] in TFA; [d] BP86D3/def2-TZVP(COSMO).
; [c] in TFA; [d] BP86D3/def2-TZVP(COSMO).
The CVs for the di-substituted complex 2 display two irreversible reduction waves at Epc=−2.10 and −2.51 V while only one irreversible reduction peak was observed at −2.02 V for complex 4 (Figure S10, see Supporting Information). The potentials have shifted to more negative values in comparison to 1 and 3 due to the presence of two phosphine ligands. The cyclic voltammograms for complexes 1–4 were also measured at different scan rates (50-1000 mV s−1); the peak current of the reduction waves was proportional to the square root of the scan rate thus, indicating that the electrochemical processes were diffusion-controlled (Figure S11, see Supporting Information).61
The calculated reduction potentials of the complexes 1–4 relative to Fc0/+ match very well with the corresponding experimental values as reported in Table 1. B3LYP results are given in the Supporting Information and are deviating more from experimental values (Table S7). The reduction potential becomes more negative with the replacement of CO ligands with σ-donor phosphine ligands. Therefore, for the di-substituted complexes (2 and 4), the first one-electron reduction occurs at more negative potentials as compared to that for the mono-substituted counterparts 1 and 3 (see Table 1).
In the mono-reduced pdt-bridged complexes 1− and 2−, the unpaired spin density is delocalized over the Fe metal atoms, while in the reduced bdt-bridged complexes 3− and 4−, the spin density is localized on one Fe atom only.
It is noteworthy that, upon reduction of the pdt-bridged complexes 1 and 2, all Fe−S bonds of the reduced species 1− and 2−, increase in length (2.28/2.35 Å in 1/1− and 2.29/2.33 Å in 2/2−) but remain intact. The Fe−Fe bond length slightly changes from 2.55 Å in 1 to 2.87 Å in 1− and from 2.54 Å in 2 to 2.67 Å in 2−.
In the reduced bdt-bridged species 3− and 4−, however, one of the Fe−S bonds breaks and Fe⋅⋅⋅S distances increase significantly (from 2.31 Å to 3.06 Å (in 3→3−); from 2.32 Å to 3.60 Å (in 4→4−)). This opening provides an accessible site for protonation (see below). However, no significant changes are observed in the Fe−Fe bond distances (2.51 Å to 2.56 Å in 3/3− and 2.50 Å to 2.58 Å in 4/4−).
It is worth mentioning that the unpaired electron spin density in 3− and 4− is only localized at the Fe atom with the open coordination site. Since the aromatic bdt-bridged reduced species (3− and 4−) possess an accessible coordination site, proton reduction would be easier in complexes 3 and 4 as compared to that in complexes 1 and 2. The Gibbs free energy change (ΔG) for the first one-electron reduction (E) of 1/1− (−298 kJ mol−1) and 2/2− (−244 kJ mol−1) is less negative than that of 3/3− (−318 kJ mol−1) and 4/4− (−290 kJ mol−1). We can thus discriminate the effect of mono- versus di-substitution (+54 kJ mol−1 for pdt and +28 kJ mol−1 for bdt complexes) and substitution of an alkyl for an aromatic di-thiolate ligand (−20 kJ mol−1 for mono- and −46 kJ mol−1 upon di-substitution).
Also, the calculated first one-electron reduction potentials of the bdt-bridged complexes 3 (−1.60 V) and 4 (−1.90 V) are less negative than for the pdt-bridged complexes 1 (−1.81 V) and 2 (−2.37 V). It is interesting to note that the second one-electron reductions of 3−/32− (−1.77 V) and 4−/42− (−2.09 V) also occur at a low potential, while for the pdt-bridged complexes 1−/12− (−2.46 V) and 2−/22− (−2.60 V), they appear to occur at higher potentials.
The close first and second one-electron redox potentials in 3 and 4 may actually make a clear experimental assignment difficult. Therefore, for the hydrogen evolution reaction (HER), we investigate the EECC and ECEC reaction mechanisms for complexes 3 and 4, while for complexes 1 and 2 only the ECEC reaction mechanism is proposed (where E=Electrochemical and C=Chemical).
Cyclic Voltammetry in Presence of Proton SourceComplexes 1–4 were examined with regard to their performance as electrocatalysts for the reduction of protons to molecular hydrogen in the presence of three different acids (acetic, trifluoroacetic (TFA) and perchloric acid (HClO4)). The pdt-bridged complexes 1 and 2 show no catalytic activity in the presence of the weak acetic acid due to the high pKa value of the acid in comparison to the one-electron reduced species.62
CVs of 3 in the presence of acetic acid display a new peak at −2.12 V versus Fc/Fc+, which shifts cathodically with the increase in the amount of acid (Figure 2). In addition, a second peak is observed at −2.43 V. The new peaks appear at a value more negative than the reduction of the complex 3 at −1.65 V in the absence of acid. The peak at −2.12 V disappears completely after adding 354 mm of acid, while the second peak at −2.43 V persists at higher concentrations of acid in solution.

Cyclic voltammograms for complex 3 (1.11 mm) in acetonitrile without acid (– -) and with increasing amounts (14–354 mm) of acetic acid (—) at a scan rate of 0.1 V s−1.
The increase in current paralleling the increasing amount of acid can be attributed to the reduction of protons yielding molecular hydrogen.58b For complex 4, a peak is first observed at −2.46 V with the appearance of a second peak at −2.36 V upon adding 28 mm of acid, which shifts cathodically with the increase of acid concentration in the solution (Figure 3). Subsequently, only one peak remains up to 270 mm of acid in the solution. This indicates that two catalytic reaction mechanisms may be occurring simultaneously, that is, an ECEC and an EECC mechanism.

Cyclic voltammograms for complex 4 (1.13 mm) in acetonitrile without acid (– -) and with increasing amounts (14–270 mm) of acetic acid (—) at a scan rate of 0.1 V s−1.
The CVs were corrected for the background currents without catalyst in the presence of acetic acid. (see Supporting Information, Figure S12). Hence, the acid-induced currents in the presence of catalysts can be attributed to catalytic turnover.63 From the plot of peak currents (icat) versus acid concentration for complexes 3 and 4, it can be seen that the bdt-bridged di-substituted complex 4 is a slightly better catalyst than the mono-substituted complex 3 in the presence of acetic acid (Figure 4 and Figure S13, Supporting Information). However, the overpotential for 3 (0.66 V) is lower than that of 4 (0.90 V).

Plots of icat/μA vs. [CH3COOH]/m for 3 (1.11 mm) (▪) and 4 (1.13 mm) (•) for the first reduction peak at a scan rate of 0.1 V s−1. The negative sign for the current has been ignored.
Electrochemical investigations for the four complexes 1–4 were also performed in the presence of trifluoroacetic acid (TFA). A similar pattern in the CVs can be observed for all the four complexes in the presence of TFA (Figure 5 and Figures S14–S16, Supporting Information). The reduction potentials of the bdt-bridged complexes in presence of TFA are lower than those of the aliphatic ones, hence leading to lower overpotentials. Moreover, the presence of two phosphine ligands further lowers the overpotential for the di-substituted complexes.

Cyclic voltammograms for complex 2 (0.67 mm) in acetonitrile without acid (– -) and with increasing amounts (1.2–54.5 mm) of TFA (—) at a scan rate of 0.1 V s−1. Reverse scans have been omitted for clarity.
As can be seen from the current versus acid concentration plots, mono-substituted complexes 1 and 3 display comparable catalytic currents (Figure 6). However, in the case of di-substituted complexes, higher currents are observed for the bdt-bridged complex 4 in comparison to the pdt-bridged complex 2 (Figure 6).

Plots of icat/μA vs. [TFA]/m for the first reduction peak of 1 (▪), 3 (•), 2 (▪) and 4 (•) at a scan rate of 0.1 V s−1. The negative sign for the current has been ignored.
In the presence of HClO4, complexes 2 and 4 showed higher currents than 1 and 3 as shown in Figure S17 (see Supporting Information). Furthermore, the observed higher currents for the aromatic (bdt) dithiolate complexes (3 and 4) are higher compared to the aliphatic (pdt) systems (1 and 2). Therefore, we can say that the bdt-bridged complexes are better catalysts than the corresponding pdt-bridged complexes. Strong acids (pKa <10) are able to protonate the reduced intermediates more easily than weak acids. Moreover, due to the aromatic nature of the thiolate ligand in 3 and 4, the one-/two-electron-reduced species are easier to protonate in comparison to the intermediates formed from the pdt-bridged complexes.
Comments (0)