
Remember me


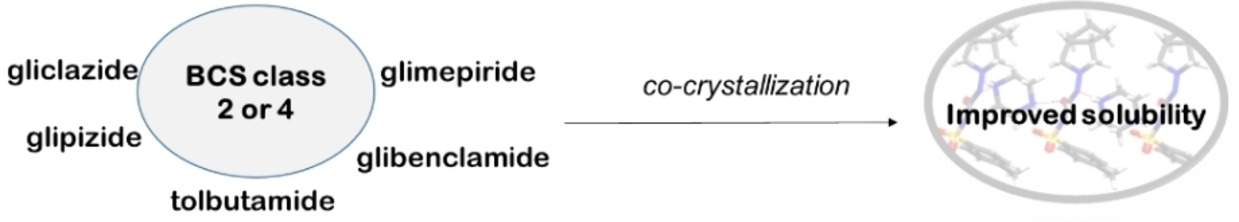
Even though enormous effort and capital are spent on discovering and developing new drugs,1, 2 the successful candidates often show poor physicochemical properties, i. e., solubility, stability, dissolution rate, etc.3 Solubility is a fundamental property that affects the bioavailability and dosage of drugs.4 Because more than 40 % of new chemical entities (NCE) are insoluble in water,5 there has been an increasing interest in designing strategies that can enhance the solubility of drug molecules without changing their molecular structure and activity.
Crystal engineering, which is “the understanding of intermolecular interactions in the context of crystal packing and the utilization of such understanding in the design of new solids with desired physical and chemical properties”,6 presents an opportunity to improve the physical properties of active pharmaceutical ingredients (APIs). In general, the physical properties of a compound are attributable to the molecular arrangements and the intermolecular interactions between the molecules.7 Therefore, manipulating these factors through the formation of salts and co-crystals can improve the properties of the parent API.7 A new multicomponent crystal with unique or enhanced physical properties can be obtained by co-crystallizing an API with a pharmaceutically acceptable compound (a coformer). The new multicomponent crystal is known as a pharmaceutical co-crystal or salt. The coformer does not interfere with the activity of the API but merely enhances certain of its physical properties.7
Diabetes mellitus is a group of diseases, namely Type 1, Type 2, and gestational diabetes. Even though not curable, the diseases can be managed using appropriate medications. Some of the drugs used for managing Type 2 diabetes have low aqueous solubility and are thus classified as either class 2 or class 4 under the biopharmaceutics classification system (BCS).8 BCS is a system for grouping drugs according to their aqueous solubility and gastrointestinal permeability.8 There are four classes of drugs according to this classification, viz Class 1: high solubility – high permeability, Class 2: low solubility – high permeability, Class 3: high solubility – low permeability, and Class 4: low solubility – low permeability.8 Several researchers have dedicated their efforts to designing and preparing pharmaceutical co-crystals that can improve the solubility of class 2 and class 4 drugs. In general drug development, the co-crystal approach has achieved relative success wherein several drugs now have improved physicochemical properties and are currently approved by the US Food and Drug Administration (FDA) as pharmaceutical co-crystals.9 A significant number of antidiabetic drugs such as gliclazide, glimepiride, glipizide, repaglinide, pioglitazone, glibenclamide and rosiglitazone fall under either BCS class 2 or class 4. Therefore, this paper aims to determine the extent to which the co-crystallization approach has been used to improve the solubility of some of these poorly soluble antidiabetic drugs. The paper will first briefly discuss the synthesis and characterization methods of co-crystals and the crystal engineering techniques that enhance solubility, followed by a review of some of the pharmaceutical co-crystals of tolbutamide, gliclazide, metformin, glipizide, glimepiride, and glibenclamide published to date. The structures of the drug molecules reviewed in this paper are shown in Figure 1.

Structures of the antidiabetic drug molecules reviewed in this paper.
2 Synthesis and Characterization of Co-crystalsThe design of pharmaceutical co-crystals involves selecting suitable coformers with functional groups capable of interacting with those of the API. This selection is usually guided by the ′synthon approach′ as well as a search on the Cambridge Structural Database (CSD)10 to determine the occurrence of a certain synthon. Some of the synthons used in the design and preparation of pharmaceutical co-crystals include pyridyl⋅⋅⋅acid, amide⋅⋅⋅acid, pyridyl⋅⋅⋅hydroxyl and amide⋅⋅⋅pyridyl heterosynthons (Figure 2). Coformers are non-toxic and are selected from the ‘Generally Regarded as Safe’ (GRAS) list approved by the FDA.11 There are, however, some challenges associated with the co-crystal approach. Even though the API and the coformer may possess complementary functional groups, co-crystal formation may also depend on the crystallization method, the solvent used, temperature, etc., and is, therefore, not always guaranteed. In addition, the inability to predict the structure of a co-crystal from its building blocks remains the greatest challenge of the co-crystal approach and crystal engineering in general. Despite these challenges, a lot of research effort has focussed on preparing pharmaceutical co-crystals of APIs, and this research is summarized in several reviews.12-17

Some of the supramolecular synthons used in the design and preparation of pharmaceutical co-crystals; (i) pyridyl⋅⋅⋅acid, (ii) pyridyl⋅⋅⋅hydroxyl, (iii) amide⋅⋅⋅pyridyl and (iv) amide⋅⋅⋅acid heterosynthons.
There are a variety of techniques that can be used for the synthesis of co-crystals. These techniques can be divided into solution-based and solid-based methods. The solution-based methods include solvent evaporation, antisolvent, cooling crystallization, reaction crystallization, and slurry conversion. The solid-based methods include contact crystallization, solid-state grinding, and melting crystallization.18 For further details on these methods, the reader is referred to a recent review by Guo et al.19 Methods of characterization of co-crystals usually include a combination of X-ray diffraction studies (powder X-ray diffraction (PXRD) and single-crystal X-ray diffraction (SCXRD)), thermal analysis (thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC)) and spectroscopy (Fourier transform infrared (FTIR), solid-state NMR (ssNMR)).20
3 SolubilitySolubility may be defined quantitatively as “the concentration of solute in a saturated solution at a certain temperature’’ and qualitatively as the “spontaneous interaction of two or more substances to form a homogeneous molecular dispersion”.21 The process of solubilizing a co-crystal shown in Figure 3 involves (i) breaking bonds in a co-crystal, (ii) breaking bonds in a solvent, and (iii) formation of solvent-solute interactions.21 Solubility is influenced by the lattice energy and the solvent-solute interactions and, the formation of co-crystals has the effect of lowering the lattice energy, thereby enhancing solubility.22 Certain techniques can be utilized to enhance the solubility of a drug using the crystal engineering approach. Salt formation and coformer solubility are the widely-used techniques. The following two sections discuss these two techniques in detail.

Solubilization of a co-crystal; the process involves (i) breaking solute-solute bonds, (ii) breaking solvent-solvent bonds, and (iii) formation of solvent-solute bonds.21
3.1 Coformer SolubilityCoformer solubility is often used as an indicator for solubility improvement of co-crystals. Thus, in addition to possessing complementary functional groups, a coformer may be selected based on its solubility. A study on carbamazepine co-crystals concluded that a coformer solubility value ten times higher than the drug would yield a more soluble co-crystal than the parent drug.12 The enhanced solubility due to coformer solubility is attributed to the “decrease in the solvation barrier for a co-crystal to an extent proportional to that of the pure coformer”.23 Several experimental studies have found a correlation between coformer solubility and co-crystal solubility while some studies found a partial correlation or a negative correlation.24-27
3.2 Salt FormationPoorly soluble drugs containing ionizable groups can be formulated as salts to improve solubility.28 Thus, it is estimated that more than 50 % of drugs currently available in the market are formulated as salts.28 While noting that pKa differs in different solvents, the pKa rule can be used to predict salt formation.28 A study conducted for multicomponent crystals deposited in the CSD concluded that salt formation is expected exclusively for ΔpKa >4 (ΔpKa=pKa[protonated base]−pKa[acid]) while co-crystal formation is expected exclusively for ΔpKa <−1. In the ΔpKa range of −1 to 4, there is an almost equal chance of either co-crystal or salt formation.29 In a study by Thakuria et al., the solubility values of olanzapinium monomaleate and dimaleate salts were found to be 225 and 550 times higher than the parent drug, olanzapine, respectively.30 Another example is a study by Portell et al., where a malate salt of the drug ziprasidone achieved a 14-fold enhancement in solubility compared to the currently marketed hydrochloride salt of ziprasidone.31 Other salts of drugs reported to have higher solubility compared to the parent drug include salts of amoxapine32 and sildenafil.33
4 Multicomponent Crystals of Antidiabetic DrugsTolbutamide is a first-generation sulfonylurea drug, while glipizide, gliclazideand glibenclamide are second-generation. Gimepiride is a third generation drug. The sulfonylurea group can form interactions with various functional groups such as O−H, N−H, py−N, and COOH. This section reviews pharmaceutical co-crystals or salts of the six drug molecules shown in Figure 1.
4.1 Gliclazide and TolbutamideGliclazide (GLZ) and tolbutamide (TOL) are used to treat Type 2 diabetes by stimulating insulin secretion. Per the BCS, gliclazide and tolbutamide are class II drugs characterized by low solubility and high permeability. A search on the CSD (version 5.41, March 2020) using GLZ as the search fragment produced seven hits. Of these hits, one is a crystal structure of GLZ, four are salts of GLZ with metformin, 3,4 diaminopyridine, 4-aminopyridine, and benzamidine. The remaining two are co-crystals of GLZ with catechol and resorcinol. There are four salts of tolbutamide with n-butylamine, metformin, and two stoichiometric variations of tolbutamide and piperazine deposited on the CSD. In the GLZ crystal structure, the drug molecules interact via the N−H⋅⋅⋅O=S and N−H⋅⋅⋅O=C dimers between neighboring sulfonylurea groups (Figure 4). In the TOL structure, the drug molecules are held together by N−H⋅⋅⋅O=S and N−H⋅⋅⋅O=C interactions between the sulfonylurea groups (Figure 5).

N−H⋅⋅⋅O=S and N−H⋅⋅⋅O=C dimers between the GLZ molecules in a GLZ crystal structure (CCDC refcode SUVGUL).34

N−H⋅⋅⋅O=S and N−H⋅⋅⋅O=C hydrogen bonds between the TOL molecules in a TOL crystal structure (CCDC refcode ZZZPUS06).35
Multicomponent crystals of GLZ with catechol (CAT), resorcinol (RES), piperazine (PPZ) and p-toluene sulfonic acid (PTSA) were reported by Samie et al.36 The authors also reported two salts of tolbutamide (TOL) and piperazine in a 1 : 1 ratio (I) and a 2 : 1 ratio (II). The multicomponent crystals were characterized using DSC, FTIR, PXRD, and SCXRD. Single crystals of the GLZ−PTSA could not be obtained. The gliclazide molecules in the GLZ−CAT structure interact with each other via the N−H⋅⋅⋅O=S dimer interactions between the sulfonyl groups and they interact with the catechol molecules via the O−H⋅⋅⋅O=C(GLZ) and N−H⋅⋅⋅O=S(GLZ) hydrogen bonds. Catechol molecules interact with each other via O−H⋅⋅⋅O hydrogen bonds (Figure 6). In the GLZ−RES structure, the gliclazide molecules are held together by the N−H⋅⋅⋅O=S dimer interactions while gliclazide and resorcinol molecules are linked via O−H⋅⋅⋅O=C(GLZ) as well as N−H⋅⋅⋅O−H(RES) hydrogen bonds (Figure 7). The GLZ−PPZ salt contains N−H+⋅⋅⋅−N, N−H⋅⋅⋅O=C(GLZ) and N−H⋅⋅⋅O=S(GLZ) hydrogen bonds between the protonated piperazine and the deprotonated gliclazide (Figure 8). Unlike the GLZ−CAT and GLZ−RES structures, there are no interactions between the sulfonyl groups of the gliclazide molecules in the GLZ−PPZ structure.

Hydrogen bond interactions in the GLZ−CAT structure (CCDC refcode WAQZOG). The GLZ molecules interact with each other via the N−H⋅⋅⋅O=S dimer between the sulfonyl groups, and they interact with the catechol molecules via the O−H⋅⋅⋅O=C(GLZ) and N−H⋅⋅⋅ O=S(GLZ) hydrogen bonds.36

Hydrogen bond interactions in the GLZ−RES crystal structure (CCDC refcode WAQZUM). GLZ molecules interact via the N−H⋅⋅⋅ O=S dimer while GLZ and RES are linked via O−H⋅⋅⋅O=C(GLZ) as well as N−H⋅⋅⋅O−H(RES) hydrogen bonds.36

Hydrogen bond interactions in the GLZ−PPZ crystal structure (CCDC refcode WARBAV). The GLZ−PPZ salt contains N−H⋅⋅⋅O=C(GLZ) and N−H⋅⋅⋅O=S(GLZ) hydrogen bonds between the protonated piperazine and the deprotonated gliclazide.36
Solubility and dissolution experiments were conducted in a pH 7.4 phosphate buffer, and were found to follow the order GLZ−PPZ>GLZ−CAT>GLZ−RES>GLZ−PTSA>GLZ (see Table 1 for solubility values). The high solubility of GLZ−PPZ was attributed to the high solubility of PPZ as well as salt formation. In both the TOL−PPZ(I) and TOL−PPZ(II) structures, the TOL and PPZ molecules interact with each via the HN−H+⋅⋅⋅−N(TOL), HN−H⋅⋅⋅O=C(TOL) and HN−H⋅⋅⋅O=S(TOL) hydrogen bonds (Figure 9 and 10). In the TOL−PPZ(I) structure the deprotonated TOL interact with each other via N−H⋅⋅⋅O=S hydrogen bonds while in the TOL−PPZ(II) structure, these interactions are absent. The solubility of the tolbutamide salts was found to follow the order TOL−PPZ(I)>TOL−PPZ(II)>TOL(API). The higher solubility values of the two salts TOL−PPZ(I) and TOL−PPZ(II) were also attributed to the high solubility of the PPZ coformer. Solubility values are also given in Table 1.
Table 1. Solubility values for the gliclazide, tolbutamide, and glipizide and their co-crystals/salts in different dissolution media.Co-crystal
Dissolution medium
Solubility mg/mL ±SD
Ref
GLZ
Phosphate buffer pH 7.4
1.170
Samie et al.
GLZ−CAT
7.113
GLZ−RES
4.113
GLZ−PTSA
3.129
GLZ−PPZ
7.710
TOL
4.179
TOL−PPZ(I)
331.896
TOL−PPZ(II)
9.987
GLZ
Phosphate buffer pH 7.4
2.10±0.2
Chadha et al.
GLZ−SA
4.49 ±0.3
GLZ−MA
4.05±0.2
GLZ
0.217±0.0687
Ibrahim et al.
GLZ−MA ph[a]
Distilled water
0.698±0.0588
GLZ−MA cp[b]
6.33±0.0588
GPZ
Phosphate buffer pH 7.4
0.19±0.02
Rani et al.
GPZ−PA
1.15±0.04
GPZ−AA
0.85±0.03
GPZ−INA
0.70±0.02
GPZ−FA
0.44±0.02
GPZ−SRA
0.32±0.03
GPZ
Distilled Water
0.00135
GPZ−OA
0.0151
Pandey et al.
GPZ−BA
0.0175
GPZ−MAL
0.0303
GPZ−SA
0.0405
[a] Physical mixture. [b] co-precipitate
Hydrogen bond interactions in the TOL−PPZ(I) crystal structure (CCDC refcode WAQMIN). TOL and PPZ interact with each other via the HN−H⋅⋅⋅N(TOL), HN−H⋅⋅⋅O=C(TOL) and HN−H⋅⋅⋅O=S (TOL) hydrogen bonds.36

Hydrogen bond interactions in the TOL−PPZ(II) structure (CCDC refcode WARBEZ). TOL and PPZ interact with each other via the HN−H⋅⋅⋅N(TOL), C=O⋅⋅⋅H2N(PPZ) and HN−H⋅⋅⋅O=S(TOL) interactions while TOL interact with each other via the N−H⋅⋅⋅O=S hydrogen bonds.36
Co-crystals of GLZ with succinic acid (SA) and malic acid (MA) were prepared by Chadha and co-workers.37 The co-crystals were synthesized using the liquid-assisted grinding method and characterized using DSC, PXRD, and FTIR. The crystal structures were determined from the PXRD data. In the GLZ-SA structure, the GLZ and SA molecules are held together by O-H⋅⋅⋅O=S(GLZ) hydrogen bonds, while in the GLZ-MA the GLZ and MA molecules interact via O−H⋅⋅⋅O=C(GLZ), C=O⋅⋅⋅N(GLZ) and S=O⋅⋅⋅O(MA) hydrogen bonds. GLZ-MA is a 3D structure, whereas GLZ-SA is 2D. The solubility and dissolution rate were measured in pH 7.4 phosphate buffer. The equilibrium solubility of the co-crystals was found to be approximately two times higher than that of GLZ and followed the order GLZ-SA>GLZ-MA>GLZ. The enhanced solubility of the co-crystals was attributed to the higher solubility values of SA and MA. When comparing GLZ-SA and GLZ-MA, the improved solubility of GLZ-SA was attributed to its 2D structure compared to the 3D structure of GLZ-MA. The solubility values are given in Table 1.
Aljohani and co-workers38 attempted to co-crystallize GLZ with antihypertensive drugs chlorothiazide (CTZ), hydrochlorothiazide (HCTZ), indapamide (IND), triamterene (TRI) and nifedipine (NIF). These antihypertensive drugs are also BCS class II. Solution crystallization and mechanochemical methods did not yield co-crystals of GLZ with any of the antihypertension drugs. However, ball milling produced co-amorphous phases of GLZ with HCTZ, TRI, IND, CTZ, and NIF. Only the co-amorphous phase of GLZ-TRI was stable in air and dissolution media and showed an enhanced dissolution rate.
Ibrahim and co-workers39 prepared a co-crystal of GLZ and malonic acid (MAL) by co-precipitation. The co-crystal formation was confirmed using PXRD, DSC, FTIR, NMR, and SEM. The solubility of the co-crystal, determined in distilled water, was found to be higher than that of pure GLZ and the physical mixture of GLZ and MAL (see Table 1 for solubility values).
Bruni et al.40 prepared a co-crystal of tromethamine with GLZ using a combination of mechanical and thermal activation methods. The co-crystal was characterized using DSC, FTIR, PXRD, NMR, and SEM-EDS. The co-crystal was found to have an enhanced solubility and dissolution rate compared to the GLZ.
Putra et al.41 reported two iso-structural salts of GLZ prepared from 4-aminopyridine (4AMP) and 3,4-diaminopyridine (3,4 DAMP). The salts were characterized using DSC, PXRD, and SCXRD. GLZ and 4AMP are held together via HN−H⋅⋅⋅O=C(GLZ), HN−H⋅⋅⋅O=S(GLZ), HN−H⋅⋅⋅N(GLZ) and N−H+⋅⋅⋅−N(GLZ) interactions in the GLZ-4AMP crystal. There are no interactions between the deprotonated GLZ (Figure 11). In the GLZ-3,4DAMP structure, GLZ and 3,4DAMP are held together via N−H+⋅⋅⋅−N(GLZ), HN−H⋅⋅⋅O=C(GLZ), HN−H⋅⋅⋅O=S(GLZ) hydrogen bonds. The GLZ molecules interact with each other via the N−H⋅⋅⋅O=S hydrogen bonds (Figure 12). The dissolution studies showed that the two salts had higher dissolution rates than pure GLZ, with the GLZ-4AMP showing the fastest dissolution rate. This was attributed to the low packing efficiency of the GLZ-4AMP salt.

Hydrogen bond interactions in the GLZ-4AMP crystal structure (CCDC refcode UYAMEO). GLZ and 4AMP interact via the HN−H⋅⋅⋅O=C(GLZ), HN−H⋅⋅⋅O8S(GLZ), HN−H⋅⋅⋅N(4AMP) and N−H+⋅⋅⋅−N(GLZ).41

Hydrogen bond interactions in the GLZ-3,4DAMP crystal structure (CCDC refcode UYALOX). GLZ and 3.4DAMP molecules interact with each other via N−H+⋅⋅⋅−N(GLZ), N−H⋅⋅⋅O=C, HN−H⋅⋅⋅O=S(GLZ). The GLZ molecules interact with each other via the N−H⋅⋅⋅O=S interactions.41
4.2 GlipizideGlipizide (GPZ) is an oral hypoglycemic drug. It is a BCS class II drug characterized by low solubility and high permeability. A search on the CSD produced only three crystals structures. Two of the crystal structures are of pure glipizide,36, 42 and one crystal structure is a glipizide-piperazine isobutanol methanol solvate hydrate salt.36 The glipizide crystal structure consists of GPZ molecules interacting via the N−H⋅⋅⋅O=S and N−H⋅⋅⋅O=C dimers. In addition, the molecules are also held together by amide⋅⋅⋅amide hydrogen bonds (Figure 13). Rani et al.43 reported co-crystals of glipizide with picolinic acid (PA), adipic acid (AA), isonicotinic acid (INA), fumaric acid (FA) and sorbic acid (SRA). The co-crystal formation was confirmed using DSC, FTIR, ssNMR, and PXRD. Crystal structures were solved from the PXRD data. The GPZ-PA structure consists of O−H⋅⋅⋅ O=C(GPZ) hydrogen bonds between the GPZ and PA. The GPZ molecules interact with each other via N−H⋅⋅⋅N hydrogen bonds. There are also intramolecular N−H⋅⋅⋅O=S hydrogen bonds on the GLZ molecules.

Hydrogen bond interactions in the GPZ crystal structure (CCDC refcode SAXFED01).36 The GPZ molecules interact via the N-H⋅⋅⋅O=S and N−H⋅⋅⋅O=C dimers of the sulfonylurea group.
The GPZ-AA structure consists of GPZ and AA molecules interacting via O−H⋅⋅⋅N(GPZ) and N−H⋅⋅⋅O=C(AA) hydrogen bonds. In the GPZ-INA structure, GPZ and INA interact via O−H⋅⋅⋅O=S(GPZ) as well as N−H⋅⋅⋅C=O(INA) hydrogen bonds. GPZ-FA structure consists of the GPZ and FA molecules interacting via O−H⋅⋅⋅N (GPZ), FA molecules interacting via the carboxylic acid dimer, and GPZ molecules interacting via N−H⋅⋅⋅O=C hydrogen bonds. The GPZ-SRA consists of SRA, and GPZ molecules held together by N−H⋅⋅⋅O=C(SRA) and O−H⋅⋅⋅S=O(GPZ) hydrogen bonds. Equilibrium solubility measurements were performed in phosphate buffer pH 7.4. The solubility values of all the co-crystals are higher than that of GPZ and they follow the order GPZ-PA (6 fold)>GPZ-AA (4,5 fold)>GPZ-INA (3,7 fold)>GPZ-FA (2,3 fold)>GPZ-SRA (1,7 fold). The enhanced solubility of GPZ-PA and GPZ-AA was attributed to the high solubility of the PA and AA coformers. The solubility order of GPZ-INA and GPZ-FA was attributed to the fact that the GPZ-FA crystal contains homomeric interactions which are difficult to break compared to the heteromeric interactions found in GPZ-INA. The solubility values are given in Table 1.
Pandey and co-workers44 reported four co-crystals prepared from glipizide and oxalic (OX), malonic (MAL), stearic (SA), and benzoic (BA) acids. The co-crystal formation was confirmed using DSC, FTIR, PXRD, and SEM. The solubility was measured in distilled water and followed the order GPZ<GPZ-OX<GPZ-BA<GPZ-MA<GPZ-SA (see Table 1). All co-crystals had higher solubility values than GPZ, with GPZ-SA displaying the most enhanced solubility. Its higher solubility was attributed to the surfactant properties of stearic acid.
4.3 Glimepiride and MetforminGlimepiride is a BCS class II drug characterized by low solubility and high permeability. Two polymorphs of the drug have been reported.45, 46 In the Form I structure, the GMP molecules are held together via N−H⋅⋅⋅O=C dimers. In addition, there are intramolecular N−H⋅⋅⋅O=C hydrogen bonds on the GMP molecules (Figure 14). In Form II, the GMP molecules are held together via N−H⋅⋅⋅O⋅⋅⋅H−N hydrogen bonds wherein C=O acts as a bifurcated hydrogen bond acceptor. Like Form I, there are also intramolecular N−H⋅⋅⋅O=C hydrogen bonds on the GMP molecules in Form II (Figure 15).

The crystal structure of GMP Form I (CCDC refcode TOHBUN01).45 The GMP molecules interact via N−H⋅⋅⋅O=C dimers.

The crystal structure of GMP Form II (CCDC refcode TOHBUN02). The GMP molecules interact via N−H⋅⋅⋅O⋅⋅⋅H−N hydrogen bonds.
Comments (0)