Safety and efficacy of genetic MECP2 supplementation in the R294X mouse model of Rett syndrome
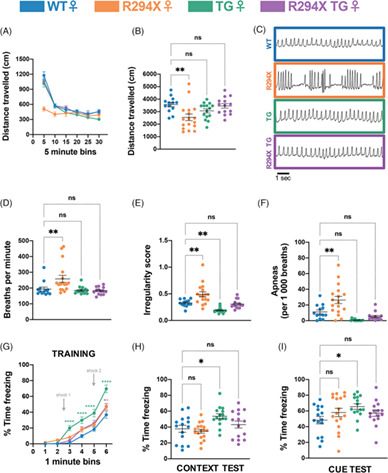
Rett syndrome is a neurodevelopmental disorder caused predominantly by loss-of-function mutations in MECP2, encoding transcriptional modulator methyl-CpG-binding protein 2 (MeCP2). Although no disease-modifying therapies exist at this time, some proposed therapeutic strategies aim to supplement the mutant allele with a wild-type allele producing typical levels of functional MeCP2, such as gene therapy. Because MECP2 is a dosage-sensitive gene, with both loss and gain of function causing disease, these approaches must achieve a narrow therapeutic window to be both safe and effective. While MeCP2 supplementation rescues RTT-like phenotypes in mouse models, the tolerable threshold of MeCP2 is not clear, particularly for partial loss-of-function mutations. We assessed the safety of genetically supplementing full-length human MeCP2 in the context of the R294X allele, a common partial loss-of-function mutation retaining DNA-binding capacity. We assessed the potential for adverse effects from MeCP2 supplementation of a partial loss-of-function mutant and the potential for dominant negative interactions between mutant and full-length MeCP2. In male hemizygous R294X mice, MeCP2 supplementation rescued RTT-like behavioral phenotypes and did not elicit behavioral evidence of excess MeCP2. In female heterozygous R294X mice, RTT-specific phenotypes were similarly rescued. However, MeCP2 supplementation led to evidence of excess MeCP2 activity in a motor coordination assay, suggesting that the underlying motor circuitry is particularly sensitive to MeCP2 dosage in females. These results show that genetic supplementation of full-length MeCP2 is safe in males and largely so females. However, careful consideration of risk for adverse motor effects may be warranted for girls and women with RTT.






Comments (0)