
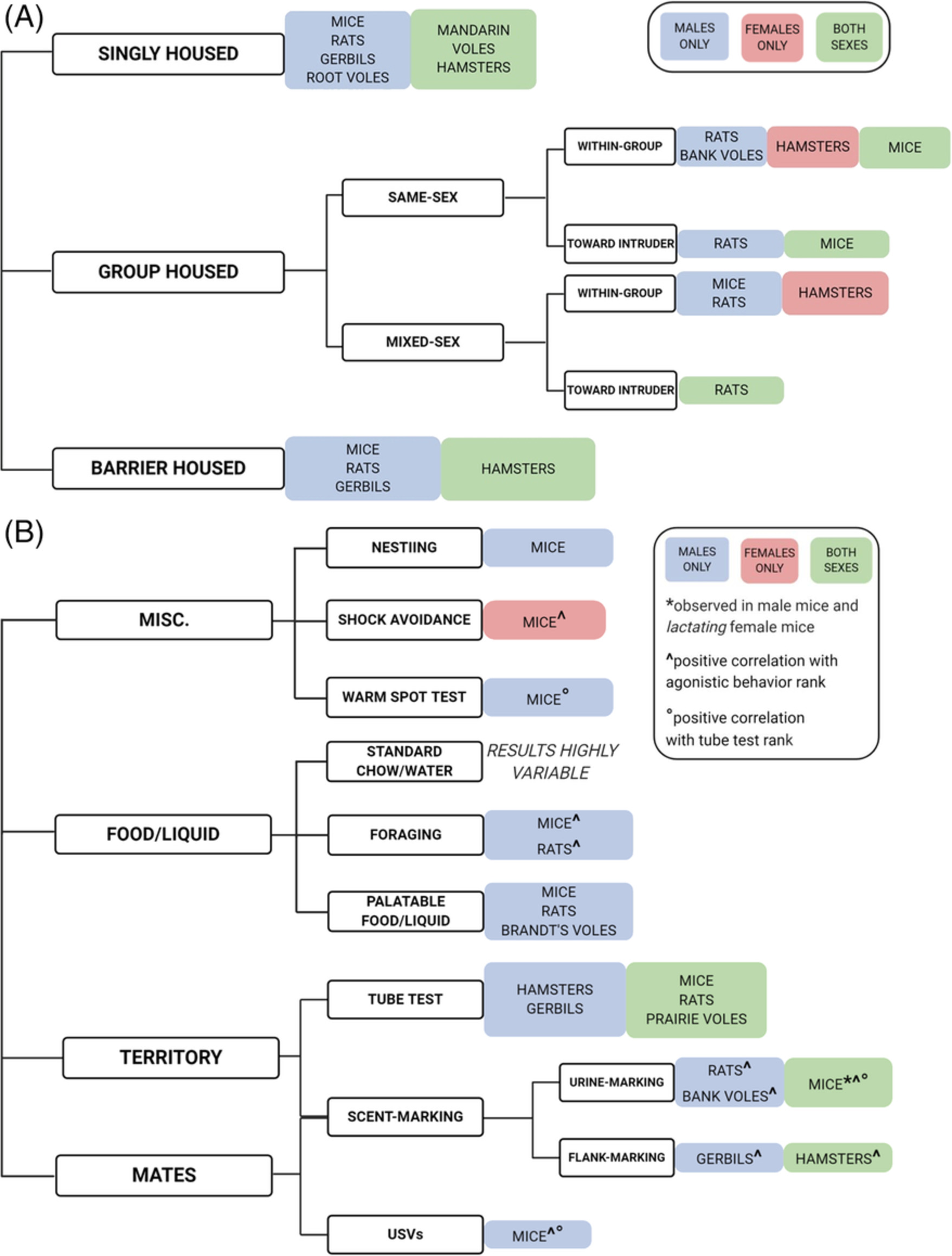
Remember me





Domesticated animals have been long known to exhibit similarities, such as: loss of pigmentation; rounded faces; smaller teeth and weaker biting force; floppy ears; shorter muzzles; curly tails; smaller cranial capacity and brain size; pedomorphosis; neotenous (juvenile) behavior; reduction of sexual dimorphism (feminization); docility; and reduced aggression and glucocorticoid levels.1-4 These changes are collectively referred to as “domestication syndrome.” A theoretical analysis by Wilkins et al.4 proposed that these common changes may be due to reduced numbers and/or delayed migration of neural crest cells during embryogenesis, that end up forming the cells that make up the aforementioned tissues.
White-rumped munia (WRM; Lonchura striata) is a songbird species belonging to Passeriformes, which was imported and brought into captivity from China to Japan around 1762 CE.5 Munias were initially used to foster exotic birds, which requires calmness under captivity and acceptability of non-kin individuals or species.6 By artificially selecting against aggressive individuals, Bengalese finches (BF; Lonchura striata var. domestica) evolved as the domesticated strain of munias; they typically have lower levels of the stress hormone corticosterone (CORT), possibly because parenting in small cages requires stress tolerance.7 The delayed migration of neural crest cells may cause the overall white appearance of the domesticated BFs,7 relative to the mostly brown plumage of munias that only have a small white patch on the rump. The assumption that the BF is a domesticated strain of the WRM is supported by the fact that F1 hybrids between BFs and WRMs are fertile,5 by the similarity of their mostly innate and sexually dimorphic distance calls,8 the occasional occurrences of munia-like morphs in BFs,9 as well as by avicultural records.6
One gene that has been proposed to play a role in domestication is oxytocin (OT).10 It is produced mainly in the supraoptic nucleus (SO) and the paraventricular nucleus (PVN) of the hypothalamus, from where it is released in the capillaries of the posterior pituitary to then be distributed peripherally, acting as a hormone, or from axon terminals of PVN neurons that innervate many other brain regions containing the OT receptor (OTR), acting as a neurotransmitter/neuromodulator.11 Traditionally, OT has been studied in reproductive contexts, including uterine contractions and milk secretion,12 while more recently it has been shown to be involved in a wider array of functions, including social bonding and stress suppression.13, 14 A potential role in domestication10, 15 has been based on findings that show different brain OT expression patterns in domesticated versus wild mice and rats16; strong purifying selection in the OT gene in domesticated placental mammals17; single nucleotide polymorphisms in OT in domestic camelid populations18; and different urinary OT content,19, 20 as well as OTR brain gene expression patterns in dogs and wolves.21
Further, OT has been shown to be involved in several facets of social cognition,22 like social communication23 and social avoidance,24 in mammals, through innervation of PVN OT neurons in the lateral septum25, 26 and the nucleus accumbens,27 among others. Although less extensively studied, OT's role in social behaviors has been also identified in avian species (we use the term OT for birds, as opposed to the commonly used mesotocin,28, 29 following the proposal for a universal vertebrate nomenclature for OT, its sister gene vasotocin (VT), and their receptors (OTR and VTRs), based on vertebrate-wide synteny results30). For example, in zebra finches OT has been shown to regulate aggression, affiliation, allopreening, flocking, and partner preference.28, 29, 31, 32 Moreover, in a recent study, blocking the OTR in juvenile zebra finches significantly reduced behaviors associated with approach and attention during song tutoring, ultimately affecting the song type the juveniles picked to learn.33 Several of these behaviors, as highlighted above, have been found to differ in BFs and WRMs, including aggression,1 neophobia,34 and song learning, with the domesticated BFs learning syntactically and phonologically more complex songs than those of the WRMs.35
We thus hypothesized that OT modifications may have played a role in the domestication of the BFs. To test this hypothesis, we genotyped and compared the OT nucleotide sequences from 10 wild munias and 11 Bengalese finches; we compared hypothalamic OT expression via in situ hybridization (ISH); and compared OT mRNA expression levels in the cerebrum and diencephalon via real time quantitative PCR (qPCR). We identified variants in both strains that lie in both the untranslated and protein-coding regions of the sequence, with all the latter giving rise to synonymous mutations. Several of these variants are located in transcription factor binding sites (TFBS), and show either a conserved or a relaxed evolutionary trend in the avian lineage, and in vertebrates in general. Although ISH in several hypothalamic nuclei did not reveal significant differences, our qPCR analyses showed that OT mRNA expression was significantly higher in the cerebrum, while significantly lower in the diencephalon of the BFs relative to WRMs. The brain region-specific differences in the expression of OT between BFs and WRMs suggest that differences in these central neuropeptide systems may influence the behaviors that pop up with the process of domestication.
2 MATERIALS AND METHODSAll procedures were conducted in accordance with: the guidelines and regulations approved by the Ethical committee of Azabu University; the Guidelines for Animal Experiments of Azabu University or of the RIKEN Animal Experiments Committee; the RIKEN BSI guidelines or the guidelines approved by the Animal Experimental Committee at the University of Tokyo, all of which are in accordance with the relevant guidelines and regulations approved by the Animal Care and Use Committee of Endemic Species Research Institute (ESRI).
2.1 Animals used for cDNA cloning and ISHThe origin and history of the animals used in this study are listed in Table S1. All animals used in this study were males, since in both strains (BFs and WRMs) only males can produce complex songs, while females are able to produce mainly innate calls. Adult male BFs (BF1-BF5; n = 5) were obtained from the breeding colony at RIKEN-BSI, Japan. These birds were not siblings, half-siblings, or progeny. Adult male WRMs (WRM1 and WRM2; n = 2) were kept for more than 1 year after they had been purchased from pet breeders or bred in our laboratory. All these birds were kept in group housing, under a 12-h light/dark cycle (lights on at 8:00) and were supplied ad libitum with mixed seeds and water. The birds were caught by hand and then decapitated between 9:00 and 12:00 am. The brains of the birds were frozen in optimum cutting temperature, embedded in medium (Sakura Finetek, Tokyo, Japan), and frozen on dry ice. Harvested brains were stored at −80°C until RNA extraction for cDNA cloning.
Using mist nets, wild WRMs (WRM3 and WRM4; n = 2) were captured in Huben (23°43′51″N, 120°37′58″E or 23°43′51″N, 120°37′51″E) Yunlin, Taiwan on March 14, 2019. After their capture, the birds were transported to the Endemic Species Research Institute (ESRI) and brought into the aviary. These two birds were killed by rapid decapitation on March 14 or 15, 2019. Their brains were dissected; the hypothalamus was harvested and placed into RNAlater (Ambion, Bicester, UK) and stored overnight at 4°C, shipped on dry ice to Azabu University and stored at −80°C until RNA extraction for cDNA cloning. In addition to these birds, ORF cloning was carried out using the total RNA from six WRMs (WRM5-WRM10) and six BFs (BF6-BF11) used in qPCR (Table S1).
Six additional BFs (BF12-BF17) and five WRMs (WRM11-WRM15) were used for ISH. Both BFs and WRMs were born at RIKEN and had been kept together with conspecifics until decapitation. The BFs' approximate ages were 120–240 pdh. The WRMs' ages were over 90 pdh. Both BFs and WRMs had reached sexual maturity. All the birds were kept under a 12-h light/dark cycle (lights on at 8:00 am) and were supplied ad libitum with mixed seeds and water. The birds were caught by hand and then decapitated between 9:00 to 12:00 am. The brains of the birds were frozen in optimum cutting temperature, embedded in medium (Sakura Finetek), and frozen on dry ice. Harvested brains were stored at −80°C. Their whole brains were used for ISH. For one WRM (WRM15), the lateral two-thirds of the right hemisphere had already been used for another study; thus, the left hemisphere and the medial one-third of the right hemisphere were used for ISH.
2.2 Animals used for qPCR and enzyme immunoassayThirteen wild male WRMs (WRM5-WRM10, WRM16-WRM22) were captured in Huben (23°43′51″N, 120°37′58″E or 23°43′51″N, 120°37′51″E) or Yuchi village (23°53′31″N, 120°53′31″E), in Yunlin, Taiwan between March 12 and 14, 2019. After capture, they were transported to ESRI, were brought into the aviary and were randomly assigned into cages in one room. The birds were kept for at least 1 week after they had been captured from the wild. Because capturing and transporting the birds would impose more stress on wild-captured WRMs, 1 week of accommodation under laboratory conditions was provided to minimize the stress effects. The birds were kept in group housing and on the natural photoperiod (day length was 12 h 23 min; sunrise and sunset occur at 5:24 and 18:11, respectively) and were supplied ad libitum with mixed seeds and water. They sang actively, indicating that they were not over stressed by this time. In order to reduce stimuli disparities between subjects, the day before they were decapitated, their cages were shaded with cardboard from 18:00. Adding shades (cardboard) to the birds' cages is a common practice in birdsong experiments to ensure that subjects have been exposed to the same social stimuli before decapitation. Additionally, when WRMs and BFs are kept in the dark, they do not sing. In the period between March 20 and 22, all birds were killed by rapid decapitation between 8:00 and 9:00. They weighed from 9.3 to 12 g (mean ± SD: 10 ± 0.64 g), and observation of their large and mature testes after decapitation suggested they were sexually mature males. The brains of the birds were frozen in optimum cutting temperature and embedded in medium (Sakura Finetek) on dry ice. Harvested brains were stored at −80°C, shipped on dry ice to Azabu University and stored at −80°C until RNA (WRM5-WRM10; n = 6) and peptide (WRM16-WRM22; n = 7) extraction. Six male BFs were used for qPCR (BF6-BF11). These birds were obtained from the breeding colony of the University of Tokyo, Japan and were around 2 years old (phd 506–794). The University of Tokyo population is simply a subpopulation of RIKEN colony. All six of them were placed in one cage. They were kept under a 14-h light/10-h dark cycle (lights on at 8:00) and were supplied ad libitum with mixed seeds and water. They also sang actively. In order to keep the same conditions as in Taiwan, the day before they were decapitated, we covered their cages with cardboard from 18:00 onwards. They were killed by rapid decapitation between 8:00 and 9:00. Their brains were frozen on dry ice and stored at −80°C until RNA extraction. Frozen brains were dissected into the cerebrum, diencephalon, midbrain, and cerebellum immediately before RNA extraction. The cerebellum was dissected out. The diencephalon was dissected by two coronal cuts at the level of the tractus septopalliomesencephalicus (rostral edge of the preoptic area) and the oculomotor nerves (caudal edge of hypothalamus), one parasagittal cut placed 2 mm lateral to the midline and one horizontal cut 5 mm above the floor of the brain, including the hypothalamus. The optic tectum was collected as the midbrain. The posterior telencephalon including the septum and the bed nucleus of the stria terminalis (BNST), dorsal to the anterior commissure36 and was collected as the cerebrum.
Eight BFs were prepared for enzyme immunoassay (EIA). They were kept for 19 days at the aviary of Azabu University, Japan, after they had been purchased from a pet breeder. They were around 2 years old and sang actively. Four birds were placed in one cage, kept under a 12-h light/dark cycle (lights on at 6:00) and were supplied ad libitum with mixed seeds and water. The day before they were decapitated, their cages had been covered with shaded cloth since 18:00. One bird was omitted because of cataracts. Seven birds (BF18-BF24) were killed by rapid decapitation between 8:00 and 9:00. Their brains were dissected by two coronal cuts at the level of the tractus septopalliomesencephalicus and the oculomotor nerves, one parasagittal cut placed 2 mm lateral to the midline and one horizontal cut 7 mm above the floor of the brain, including the hypothalamus and the BNST. Harvested brain tissue blocks were stored at −80°C until peptide extraction for EIAs.
2.3 Molecular cloning of songbird OT cDNATotal RNA was extracted from the hypothalamus of the BFs and WRMs; it was purified using the RNeasy Lipid Tissue mini kit (Qiagen, Hilden, Germany). The unknown cDNA sequences, including 3′- and 5′-untranslated regions (UTRs) of these two songbirds' OT genes, were analyzed by PCR amplification of 3′- and 5′-RACE fragments. The first-strand cDNA for 3′-RACE was prepared by incubation of total RNA with adaptor-oligo(dT)18 primers and SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA) at 50°C for 50 min. The PCR primers listed in Table S2 were designed based on predicted sequences retrieved from the zebra finch genome assembly (Taeniopygia_guttata-3.2.4, July 200830) and the sequence obtained from RACE fragments (Figure S1). The RACE fragments were amplified using two rounds of PCR with gene-specific primers and adaptor primers. PCR amplification of 3′-RACE fragments for OT consisted of initial denaturation at 94°C for 5 min; followed by 25 cycles of 30 s at 94°C, 30 s at 55°C, and 1 min at 72°C; and final elongation at 72°C for 5 min. The reaction was performed in a 25-μl mixture containing 0.3 μg cDNA, 0.2 mM dNTPs, 0.4 μM each forward and reverse primers, and 2.5 U Ex Taq polymerase with its buffer (TaKaRa, Shiga, Japan). The template cDNA was reverse transcribed with 5′-RACE RT primer and SuperScript III reverse transcriptase (Invitrogen), followed by poly(A) tailing of the cDNA with dATP and terminal transferase (Roche Diagnostics, Rotkreuz, Switzerland). PCR amplification of 5′-RACE fragments for OT consisted of initial denaturation at 98°C for 5 min; followed by 30 cycles of 30 s at 98°C, 30 s at 55°C, and 1 min at 72°C; and a final elongation at 72°C for 5 min. The reaction was performed in a 25-μl mixture comprising 150 ng cDNA, 0.4 mM dNTPs, 0.4 μM each forward and reverse primers, and 2.5 U Ex Taq polymerase with its buffer.
To confirm the ORF sequence, the PCR reaction was performed in a 25-μl mixture containing 0.4 mM dNTPs, 0.4 μM each forward and reverse primers (Figure S1), and 2.5 U Ex Taq polymerase with its buffer, as well as 250 ng cDNA that had been reverse transcribed with SuperScript IV VILO Master Mix (Invitrogen). PCR reaction conditions were as follows: 98°C for 1 min; 30 cycles of 98°C for 10 s, 55°C for 30 s, 72°C for 1 min, and 72°C for 5 min.
PCR products were subcloned into pGEM-T easy vectors (Promega, Madison, WI). The resultant plasmids were sequenced commercially (Fasmac, Atsugi, Japan). The ORF sequences obtained were submitted to DDBJ/EMBL/GenBank with accession numbers LC489419 and LC489420. We additionally used the genomic sequences of the OT we found in the publicly available scaffold-level and chromosome-level BF genome assemblies (LonStrDom1; RefSeq assembly accession: GCF_002197715.1; LonStrDom2, GCF_005870125.1; sequences shared in our Github).
2.4 Database analysisA putative signal peptide was predicted by using SignalP 3.0 (http://www.cbs.dtu.dk/services/SignalP/). The chromosomal location and strand orientation of the identified BF genes were determined using the Genome Data Viewer (https://www.ncbi.nlm.nih.gov/genome/gdv/). The genomic structure of the BF OT was predicted using the BF genome (lonStrDom2). Genomic regions surrounding OT were compared among human, rat, and BF using the Genome Data Viewer to analyze the synteny relationship.
For the TFBS analysis, zPicture (https://zpicture.dcode.org/) and rVista (https://rvista.dcode.org/cgi-bin/rVA.cgi?rID=zpr09032019035446775) were used. zPicture alignments can be automatically submitted to rVista 2.0 to identify conserved TFBS. RVista excludes up to 95% false-positive TFBS predictions, while maintaining a high search sensitivity. In this analysis, we used the OT nucleotide sequences of BF1 and WRM1 (Table S3), as well as the zebra finch (bTaeGut1.4.pri) and chicken OT sequences (GRCg7b) from their publicly available genomes, generated by the Vertebrate Genomes Project.37
To test for possible functional effects of the SNPs we identified, we used the Variant effect predictor tool, available in Ensembl (v. 100), for the respective sites in both the BF (LonStrDom1) and the chicken genomes (GRCg6a). Further, we used PhyloP (phyloP77way), available in the UCSC Browser, that scores the measure of evolutionary conservation at individual sites, by aligning 77 vertebrate species' genomes. The scores are compared to the evolution that is expected under neutral drift, so that positive scores measure conservation, which is slower evolution than expected, while negative scores measure acceleration, which is faster evolution than expected.38 Using this measure, we can infer whether some elements are functional, based on the rationale used widely in comparative genomics that functional genomic elements evolve more slowly than neutral sequences.39
We aligned our genotyped OT nucleotide sequences from 11 BFs and 10 WRMs using CLUSTAL W (1.81), and visualized via JalView (2.11.1.0) (alignment file in our Github; variant calling results in Table S4). We also aligned the publicly available BF OT sequence (BF 12; LonStrDom2; accession GCF_005870125.1) with the variant sites we identified. We also ran a multialignment, using the same tools, with the OT sequence of the following 29 avian species: BF, Zebra finch, Gouldian finch, Small tree finch, Medium ground-finch, Dark-eyed junco, White-throated sparrow, Blue tit, Rufous-capped babbler, Silver-eye, Flycatcher, Blue-crowned manakin, Eurasian sparrowhawk, Ruff, Spoon-billed sandpiper, Pink-footed goose, Swan goose, Golden pheasant, Ring-necked pheasant, Chicken, Indian peafowl, Turkey, Japanese quail, Helmeted guineafowl, Great spotted kiwi, Little spotted kiwi, Okarito brown kiwi, African ostrich, and Chilean tinamou (for the IDs of the genomes used, Gene-IDs and locations used for the alignment: Table S5; alignment files can be found in Github). We translated all the sequences to their respective protein sequence to decipher if the variation we identified in the exons gives rise to synonymous or non-synonymous mutations using TranslatorX (protein sequences and alignments can be found in Github)40 .
Further, a Maximum Likelihood phylogenetic amino acid tree was constructed for OT in all the avian species available in Ensembl (v.100), using TreeFAM and TreeBeST5 pipeline in the Ensembl “Gene tree” tool package (https://www.ensembl.org/info/genome/compara/homology_method.html).
To test for intraspecies variation of these sites in other avian species, we used the dbSNP (release 150) available for chicken (remapped to GRCg6a), dbSNP (release 139) available for turkey (Turkey_2.01), and dbSNP (release 148) available for zebra finch (bTaeGut1_v1.p).
2.5 In situ hybridizationWe designed antisense probes to bind to different variants of the OT mRNA. Antisense probes for the OT mRNA were synthesized from the BF OT cDNA (Figure S1 and Table S2). Corresponding sense probes were also synthesized for control ISH (Figure S2). BF OT cDNA fragments were inserted into the pGEM-T easy vectors (Promega). The plasmids were digested with restriction enzymes (NcoI or SpeI) to release the fragment; probes were synthesized using SP6 or T7 RNA polymerase (Roche Diagnostics) with digoxigenin-labeling mix (Roche Diagnostics).
Frozen brains were cut in 20-μm coronal sections on a cryostat (Leica Microsystems, Wetzlar, Germany). Every 13th section through the hypothalamus from each animal was mounted on 3-aminopropyltriethoxysilane-coated slides and stored at −80°C until use. ISH was performed as described previously,41 except that sections were postfixed in 4% paraformaldehyde for 10 min, proteinase K treatment was omitted and color development of alkaline phosphatase activity was carried out for 1 h with nitro blue tetrazolium chloride (Roche Diagnostics) and 5-bromo-4-chloro-3-indolyl phosphate (Roche Diagnostics) in detection buffer. Images of sections were captured with a LeicaMC170 HD camera attached to a Leica DM500 microscope (Leica Microsystems).
2.6 Analysis of OT mRNA expressing cellsThe “analyze particle” module of ImageJ (based on size = 50-infinity pixel and threshold = 0–150) was used to count the numbers of OT mRNA-expressing cells in the following regions of interest: the lateral hypothalamus (LHy) on the left side of the brain; the external subgroups of the supraoptic nucleus (SOe) on the left side of the brain; and PVN at the level of the anterior commissure on the both sides (Table S6). This limitation was due to the absence of the lateral two-thirds of the right brain hemisphere of a WRM. The sum of cell numbers in each area was calculated.
2.7 Statistical analyses for OT mRNA expressing cellsResults are shown as means ± standard errors of the mean and coefficients of variation (Table S6). Statistical analyses were conducted with Prism 4.0 (GraphPad Software, USA), using unpaired Student's t-tests. P values <0.05 were considered statistically significant.
2.8 Real-time qPCRWe isolated RNA from four different brain regions: cerebrum, diencephalon, midbrain, and cerebellum. Total RNA was extracted using QIAzol Lysis Reagent and column purified (RNeasy Lipid Tissue Mini Kit: Qiagen). We performed reverse transcription using Superscript First-Strand Synthesis (Invitrogen) with oligo (dT) primer. Primers for OT were designed using sequences downloaded from GenBank or Ensembl (Table S7), and for the OTR were designed based on previous studies.42, 43 The control gene was peptidylprolyl isomerase A (PPIA), which has been evaluated in two songbird species (zebra finch and white-throated sparrow) as highly stable in the brain.44 qPCR was performed using Roche LightCycler 96 System with TB Green Premix Ex Taq II (Takara), in triplicate for each sample on 96-well plate. We calculated crossing point values (CT) using the Abs Quant/2nd Derivative Max method using LightCycler 96 SW 1.1 software. CT was used to calculate ΔΔCT ([CT target gene–CTcontrol gene] − [CT target gene–CTcontrol gene] calibrator). We picked the average CT of BF samples as a calibrator. Relative expression levels between the species were calculated using the 2−ΔΔCT method45 (Tables S8 and S9) Using the value of the 2−ΔΔCT, we also analyzed our qPCR results in comparison with each specific variant we identified in our multialignments (Table S10).
2.9 Production of antiserum to the avian OTThe antiserum was raised in a rabbit immunized five times with synthetic polypeptide of the avian OT sequence (CYIQNCPIG-amide) as antigen every 1 week. The specificity of the serum was tested by a dot immunoblot assay, with the OT and its ortholog and paralog (vasotocin) in other species (OT, isotocin, vasotocin, vasopressin), where we confirmed it binds solely to avian and fish OT (also called, isotocin). Aliquots of the orthologous (oxytocin, mesotocin, isotocin) and paralogous (vasotocin and vasopressin) peptides were spotted onto a 0.2-μm polyvinylidene fluoride membrane (Immun-Blot PVDF Membrane for Protein Blotting, BIO-RAD). The membrane was air dried at room temperature and was washed for 10 min in 0.05 M Tris buffer (pH 7.6) with 0.1% Tween 20 and 0.15 M NaCl (TBS-T) and incubated for 60 min in blocking solution containing 5% skim milk in TBS-T. After blockage, the membrane was exposed for 120 min to oxytocin antiserum (1:1000 dilution in blocking buffer). After the primary immunoreaction, the membrane was further incubated with anti-rabbit biotinylated IgG secondary antibody (Agilent Technologies, Inc., Santa Clara, CA; 1:200 dilution in blocking buffer) for 120 min and avidin–biotin complex reagent (Vector Laboratories, Inc., Burlingame, CA) for 120 min, and subsequently with ImmPACT DAB peroxidase substrate solution (Vector Laboratories) for 5 min at room temperature.
2.10 OT EIAThe frozen brain tissue blocks of WRM (WRM16-22; n = 7) were dissected as described above immediately before peptide extraction. The frozen brain tissue blocks of WRM and BF were boiled for 8 min and homogenized in 5% acetic acid using a TissueLyser LT (Qiagen) for 6 min at 50 Hz. The homogenate was centrifuged at 14,000 rpm for 30 min at 4°C. The supernatant was collected and forced through a disposable C-18 cartridge (Sep-Pak Vac 1cc; Waters, Milford, MA). The retained material was then eluted with 60% methanol. The pooled eluate was concentrated in a vacuum evaporator, passed through disposable Ultrafree-MC centrifugal filter units (Millipore, Billerica, MA) and dried. The dried material was reconstituted in 220 μL Dilution buffer, and 100 μL of the sample was used for EIA at duplicate.
The samples were subjected to competitive EIA by using the antiserum described above. In brief, different concentrations of OT (0.01–100 pmol) and adjusted tissue and plasma extracts were added with the antiserum against OT (1:1000 dilution) to each antigen-coated well of a 96-well microplate (F96 maxisorp nunc-immuno plate; Thermo Fisher Scientific, Roskilde, Denmark) and incubated overnight at 4°C. After the reaction with alkaline phosphatase-labeled goat anti-rabbit IgG (Sigma; 1:1000 dilution in dilution buffer), immunoreactive products were obtained in a substrate solution of p-nitrophenyl phosphate (SIGMA FAST™ p-nitrophenyl phosphate tablet set) for 90 min, then, 20 μL 5 N NaOH solution was added to and the absorbance was read at 405 nm with a reference filter of 620 nm by iMark Microplate Reader S/N 20255 (Bio-Rad, USA; Table S11).
2.11 Statistical analyses for qPCR and EIAFor analysis of the qPCR results, we performed Steel-Dwass test (nonparametric) for multiple comparison using statistical software R (http://www.r-project.org/; R Core Team, 2013; R Foundation for Statistical Computing) to investigate the difference of relative gene expression levels between BF and WRM. And we performed unpaired Student's t-tests using Prism 4.0 (GraphPad Software) to examine the effect of the single nucleotide variants we found on OT mRNA expression in the brain of BFs and WRMs. For analysis of the EIA results, statistical analyses were conducted with Prism 4.0 (GraphPad Software), using F-tests, and unpaired Student's t-tests with Welch's correction. P values <0.05 were considered statistically significant.
3 RESULTS 3.1 Identification of OT cDNA in Bengalese finches and WRMsWe isolated OT transcripts from diencephalic tissues of 11 BFs and 10 WRMs (Figure 1A; Table S3). To confirm that our identified sequence is the BF OT-ortholog, we BLAST searched the full-length cDNA sequence against a reference BF genome assembly (lonStrDom2; accession GCF_005870125.1).47 Only one locus (BLAST hit) in the genome showed high similarity (E-value <5e−56) that was unnamed (ID: LOC110473283), located on chromosome 4 (68,957,139–68,958,557).
 Nucleotide and amino acid sequences of white-rumped munia and Bengalese finch oxytocin (OT) precursor cDNA. (A) Alignment of WRM and BF OT nucleotide sequences. Color-marked nucleotides: sites that show variation in either the BFs or the WRMs sequenced in this study; alternative nucleotides are shown marked up with the same color in close proximity. Predicted protein sequence is shown below the nucleotide sequence alignment in bold. Sequences of putative TFBS in the territory of the variant sites have been underlined with different colors; each color corresponds to the color of the fonts of the respective transcription factor. ETF: TEA domain family member 2; HIC1: HIC ZBTB transcriptional repressor 1; E2, E2F: E2F transcription factor; NERF: Ets-related factor; AP2: activating enhancer binding protein 2; AP4: activating enhancer binding protein 4; AP2GAMMA: activating enhancer binding protein 2 gamma; SP3: Sp3 transcription factor; PAX6: Paired Box 6; CHX10: C. elegans ceh-10 homeo domain-containing homolog; LHX3: LIM homeobox 3. (B) Conserved gene synteny among the human, rat, and Bengalese finch OT loci. Genes are indicated by shaded boxes. OT genes are linked by dotted lines. In rodent genomes, the gonadotropin-releasing hormone 2 gene (GnRH2) is inactivated or deleted.46 (C) Schematic representation of the Bengalese finch OT structure. Exons are boxed and numbered, and introns appear as straight black lines. Shaded and open boxes denote coding and noncoding sequences, respectively. BF, Bengalese finch; WRM, white-rumped munia
3.2 Synteny and phylogenetic analyses confirm our identified gene in BFs as the OT-ortholog
Nucleotide and amino acid sequences of white-rumped munia and Bengalese finch oxytocin (OT) precursor cDNA. (A) Alignment of WRM and BF OT nucleotide sequences. Color-marked nucleotides: sites that show variation in either the BFs or the WRMs sequenced in this study; alternative nucleotides are shown marked up with the same color in close proximity. Predicted protein sequence is shown below the nucleotide sequence alignment in bold. Sequences of putative TFBS in the territory of the variant sites have been underlined with different colors; each color corresponds to the color of the fonts of the respective transcription factor. ETF: TEA domain family member 2; HIC1: HIC ZBTB transcriptional repressor 1; E2, E2F: E2F transcription factor; NERF: Ets-related factor; AP2: activating enhancer binding protein 2; AP4: activating enhancer binding protein 4; AP2GAMMA: activating enhancer binding protein 2 gamma; SP3: Sp3 transcription factor; PAX6: Paired Box 6; CHX10: C. elegans ceh-10 homeo domain-containing homolog; LHX3: LIM homeobox 3. (B) Conserved gene synteny among the human, rat, and Bengalese finch OT loci. Genes are indicated by shaded boxes. OT genes are linked by dotted lines. In rodent genomes, the gonadotropin-releasing hormone 2 gene (GnRH2) is inactivated or deleted.46 (C) Schematic representation of the Bengalese finch OT structure. Exons are boxed and numbered, and introns appear as straight black lines. Shaded and open boxes denote coding and noncoding sequences, respectively. BF, Bengalese finch; WRM, white-rumped munia
3.2 Synteny and phylogenetic analyses confirm our identified gene in BFs as the OT-ortholog
To further confirm our identified gene in the BF was the true OT-ortholog, we ran synteny analysis on the surrounding territory of the identified gene in BF (LOC110473283). The analysis revealed that this gene is located in a genomic region that is highly conserved in tetrapod vertebrates, exactly where the OT is found in those species (Figure 1B). Conserved syntenic genes in the surrounding territory include mitochondrial antiviral signaling protein (MAVS), protein tyrosine phosphatase receptor type A (PTPRA), mitochondrial ribosomal Protein S26 (MRP26), arginine vasopressin/vasotocin (AVP/VT), fAST kinase domains 5 (FASTKD5), U-box domain containing 5 (UBOX5), and leucine zipper tumor suppressor family member 3 (LZTS3; Figure 1B). Interestingly, in the BF genome (lonStrDom2), AVP/VT was also unnamed (Gene ID: LOC110473284). Based on our synteny analysis, and on further synteny and phylogenetic results presented in Theofanopoulou et al.,48 we propose that LOC110473283 should be named oxytocin (OT) and LOC110473284 should be named vasotocin (VT) in the BF. Our phylogenetic tree using the BF putative OT in LonStrDom1 as a query sequence (ENSLSDG00000001590; unnamed) grouped this sequence with the OT-ortholog in all vertebrates (Figure S3 See Gene Tree ID: ENSGT00390000004511 for a capture of the tree only in avian species). These phylogenetic results corroborate with our synteny results in that the gene we identified in the BF is the true OT-ortholog.
Using the OT sequence from lonStrDom2 (chr4: 68,957,139-68,958,557) and our identified full-length cDNA sequence of BF OT (Figure 1A; Figure S1), we further predicted the OT genomic structure (Figure 1C). The BF OT consists of three exons, spanning 1419 base pairs. Exon 1 contains the 5′-UTR, signal peptide, OT, nonapeptide (CYIQNCPIG), and a portion of the neurophysin peptide sequence. Exon 2 and the first 48 base pairs of exon 3 contain the remaining sequence of the neurophysin peptide; the remainder of exon 3 contains the 3′-UTR (Figure 1C).
3.3 Variant calling in the BF and WRM OT cDNA and comparison with 28 avian speciesWe aligned our genotyped OT sequences from 11 BFs and 10 WRMs using CLUSTAL W (1.81; Figure 1A; alignment file in our Github; variant calling results in Table S4). We also aligned the publicly available BF OT sequence (BF12; LonStrDom2; accession GCF_005870125.1) with the variant sites we identified in our multialignment. In Figure 1A, for an easier visualization of our results, we show the alignment of two sequences that represent for each strain (WRM and BF) the alleles found in most individuals, and we note the alternative alleles next to the variant sites. We translated all the sequences to their respective protein sequence to decipher if the variation we identified in the exons gives rise to synonymous or non-synonymous mutations. Lastly, to determine if the changes we observed were specific to these strains or shared with other avian species, we aligned their sequences with OT sequences from 28 avian species (Figure 2; Figure S3 and Table S5; alignment files in Github).
 Multispecies alignment of the oxytocin (OT) nucleotide sequence in 30 avian species, with specific focus on the 10 sites that differ in BFs and WRMs. Position numbers refer to the respective positions in the alignment shown in Figure 1A. Nucleotides in black font under the alignment denote the allele present in most species in the alignment; specific percentages are given below each nucleotide. Black bars denote higher or lower conservation of the respective site in the avian species aligned. One Alignment was done using CLUSTAL W, and visualized through JalView
Multispecies alignment of the oxytocin (OT) nucleotide sequence in 30 avian species, with specific focus on the 10 sites that differ in BFs and WRMs. Position numbers refer to the respective positions in the alignment shown in Figure 1A. Nucleotides in black font under the alignment denote the allele present in most species in the alignment; specific percentages are given below each nucleotide. Black bars denote higher or lower conservation of the respective site in the avian species aligned. One Alignment was done using CLUSTAL W, and visualized through JalView
Several of the OT sequences we identified in WRMs and BFs were identical in individuals within and across strains (WRM2, WRM4, WRM6, BF6, BF9, BF10, BF11; Table S4). Variation was found in positions 1–2 (5′-UTR) of the alignment, with only 1 WRM (WRM1; 10% of the WRM sample) having CG, and the rest of the WRMs (90%) showing a deletion on this site; CG was present in five individuals (38% of the BF sample), and the rest of the BFs (62%) also had this site deleted. Interestingly, CG appeared to be the ancestral state of this site (Figure 2), with no other bird showing a similar deletion in our alignment, although the first site (C present in 66% of the birds; Figure 2) was more variable than the second (G present at a 97%; Figure 2). In positions 6–8 (5′-UTR), all WRMs, except WRM1, had GCC, while all three nucleotides were deleted in WRM1; this deletion was WRM-specific, with the BFs showing only variation on the third site in position 8, which was C/T, with C present in the 62% of our BF sample (Figure 2). GCC was the status shared by most avian species (93%, 100%, and 76%, for each site, respectively; Figure 2). To sum up, both these 5′-UTR sites were variant in both WRMs and BFs.
We identified three exonic sites (positions 62, 101, and 149) that were variant in BFs but invariant in WRMs. On the 62nd locus of the alignment (Figure 1A), all WRMs and BFs had C, except the BF4 that had T (Table S4); this site is variable in the rest of the avian species, with 55% of them having C, and the rest A, T, or G. A similar status was identified in the 101st position, where all WRMs and BFs had C, except two BFs (BF4 and BF12; Table S4) that had T, while this site was found variable in other birds (C in the 52%; Figure 2). In position 149, all WRMs and BFs had C, except for BF2 and BF7 that had G on that site, which represents the plausible ancestral state, since 93% of the avian species had G in our alignment, and C was found to be specific to the WRMs and BFs. We lastly found one exonic site (position 239) that was, in turn, variant in WRMs (G/A; G present in 80% of the WRM sample; Table S4) and invariant in BFs (G), with the majority of the avian species (83%) sharing the same allele (G; Figure 2). All of the exonic mutations identified give rise to synonymous mutations.
We found three sites in the 3′-UTR (positions 389, 400, and 468) where WRMs were variant and BFs invariant, and one site (position 432) that was, instead, variant in BFs and invariant in WRMs (Figure 2; Table S4). In position 389, only one individual (WRM 9) had A (10%), while the rest had G, as was the case for the vast majority of the avian species (97%). WRMs also showed variability (position 400: G/A; G: 80%; A: 20%) on a less conserved site across birds, where 66% of them had G. The WRM-variant site in position 468 (G/C; G: 60%; C: 40%) was, in turn, robustly conserved in the rest of the avian species (G: 93%; Figure 2). The 3′-UTR variant site in BFs (position 432: C/T; C: 62%; T: 38%) is variable across birds, with only 41% having C, and only three species, other than the BF, having T on that site.
Among the avian species we used in our alignment, some of them come from domesticated species (chicken, zebra finch, turkey, Japanese quail, and swan goose), but we were not able to find any strong pattern of convergent evolution among them. For example, although all the domesticated species in our multialignment, including the BF, have G on position 239, this G is present in 83% of the avian species, thus it cannot be considered evidence of convergent evolution.
3.4 Conserved/accelerated status of the variant sites in vertebratesFurther, we used PhyloP (phyloP77way) to measure the evolutionary conservation at the variant sites we identified, by aligning 77 vertebrate species' genomes. The sites in positions 1–2, 6–8, 62, 101, 149, and 239 gave positive scores (Suppl. File S1), which measure conservation, namely slower evolution than expected29; the sites in positions 389, 400, 432, and 468 gave negative scores, which measure acceleration, namely faster evolution than expected.38 Using this measure, we can infer whether some elements are functional, based on the rationale used widely in comparative genomics that functional genomic elements evolve more slowly than neutral sequences.39
Interestingly, the conservation status of some of these sites is different on a vertebrate-wide scale than when zoomed in an avian-specific scale. For example, the sites on positions 62 and 101 are conserved across vertebrates (Suppl. File S1), but appear to be variable in our avian multialignment (Figure 2). These same sites are variant only in BFs and not in WRMs, along with the sites on positions 8, 149, and 432, which are, in turn, conserved in both a vertebrate-wide and an avian-specific scale. These data suggest that these sites, where we found variation in BFs only, are likely to be functional and are bound to shed light on their domestication process. Especially, T in position 8, present in 38% of the BFs (Table S4) is conserved across vertebrates, and at the same time is a rare allele in our avian multialignment (present in 17% of the species; Figure 2); the same holds for T in position 432, which is also present in 38% of the BFs, is conserved in vertebrates, but it is found rarely in birds (10%).
3.5 Transcription factor-binding, functional, and variation analysesOur analysis using the zPicture and rVista softwares revealed that several of the variant sites are located in putative TFBS (Figure 1A). The site on position 62 is located close to the end of a putative binding site of the ETF (TEA domain family member 2) transcription factor, while the variant in position 101 is found in the territory of both the binding sites of the HIC1 (HIC ZBTB transcriptional repressor 1) and E2(F) transcription factors. We found both of the latter TFBS being conserved in the zebra finch. The site in position 239 was found to fall within the NERF (Ets-related factor) binding site, which was also conserved in the chicken. The two last 3′-UTR variants are located within the binding sites of several transcription factors. The site in position 432 is found in the binding sites of the following transcription factors: AP2 (activating enhancer binding protein 2), AP4 (activating enhancer binding protein 4), AP2GAMMA (activating enhancer binding protein 2 gamma), and SP3 (Sp3 transcription factor); the SP3 TFBS was also conserved in the zebra finch. Lastly, the site in position 468 is located in the PAX6 (Paired Box 6), LHX3 (LIM homeobox 3), and CHX10 (C. elegans ceh-10 homeo domain-containing homolog) TFBS, with the latter being conserved in the zebra finch.
We then used variant effect predictor tests Ensembl (v. 100) to further predict the functionality of our identified single nucleotide polymorphisms (SNPs) in the BFs and the WRMs, applied to the respective sites in both the BF (LonStrDom1) and the chicken genomes (GRCg6a), but the tests did not yield any known functional effect. We also searched for possible intraspecies variation of these sites in other avian species, using the dbSNP (release 150) available for the chicken (GRCg6a), dbSNP (release 139) available for the turkey (Turkey_2.01), and dbSNP (release 148) available for the zebra finch (bTaeGut1_v1.p), but we did not find any variation in the sites studied within any of these species.
3.6 Changes in OT mRNA expression in the cerebrum and diencephalon of BFs relative to WRMsDifferential OT gene expression in several diencephalic nuclei has been associated with social behavioral differences in several species.49 Since OT is expressed primarily in the hypothalamus,36, 50 we performed ISH for OT mRNA in BF and WRM brain sections containing several hypothalamic nuclei. BFs and WRMs had similar distributions of OT mRNA-expressing cells in the hypothalamus (Figure 3; Figure S2 for ISH with a sense and antisense OT probe), and we did not detect any significant differences in the numbers of cells expressing OT mRNA (Figure 4A–C; Table S6) in any of the nuclei tested between the two strains by ISH. These nuclei included the SOe, LHy, and PVN (Figure 3A–F). We found that the PVN contained a higher average number of OT cells in both strains (48.0 in BF; 46.7 in the WRM), compared to the SOe (7.4 in BF; 11.3 in the WRM) and the LHy (7.0 in BF; 7.6 in the WRM; Table S6).

Photomicrographs of oxytocin (OT) mRNA-expressing cells in brains of Bengalese finches and white-rumped munias. (A) and (B) SOe, external subgroups of the supraoptic nucleus. (C) and (D) LHy, lateral hypothalamic areas. (E) and (F) PVN, paraventricular nucleus; TSM, septopalliomesencephalic tract. Scale bars = 200 μm
Comments (0)