
Remember me
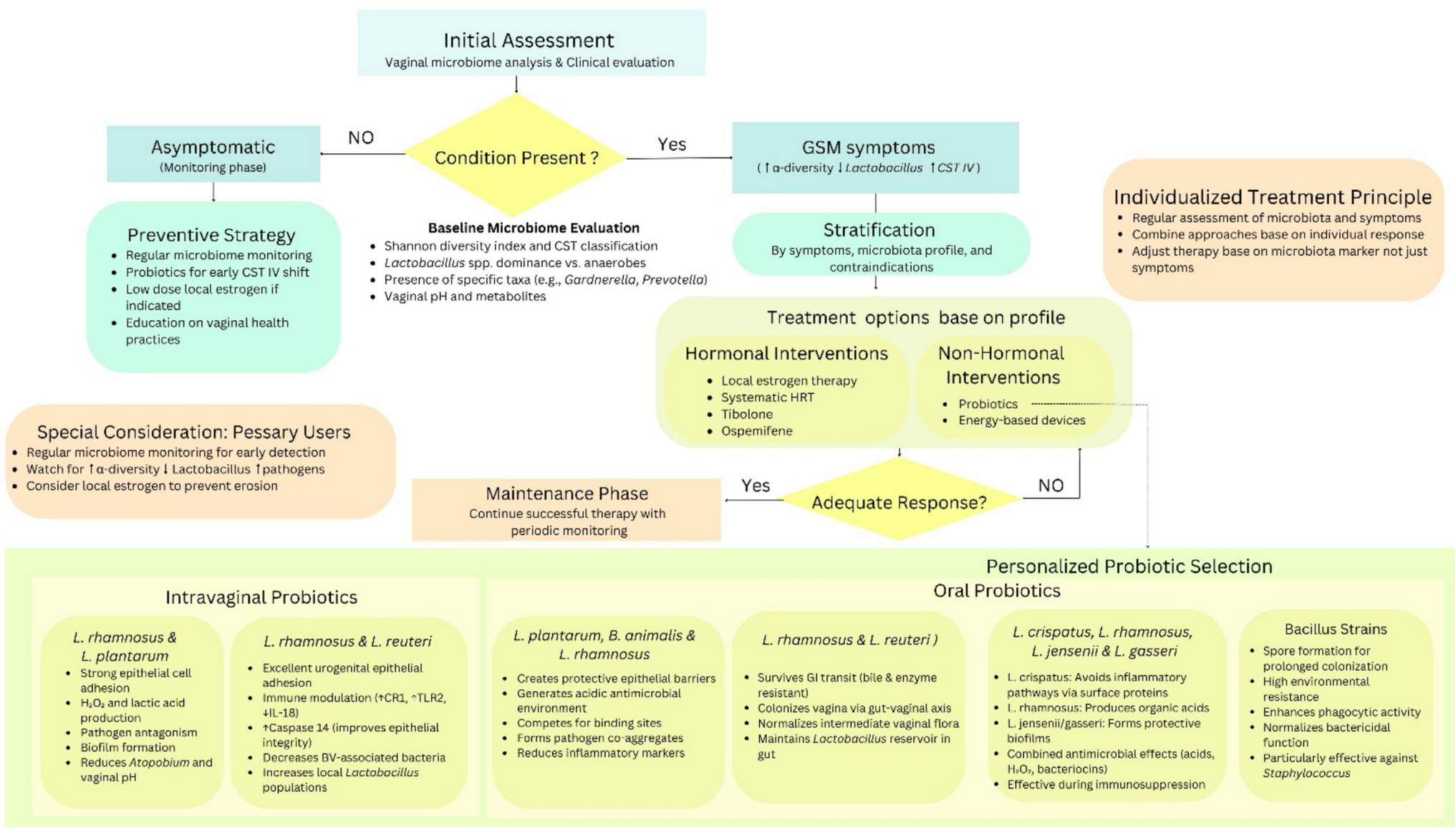
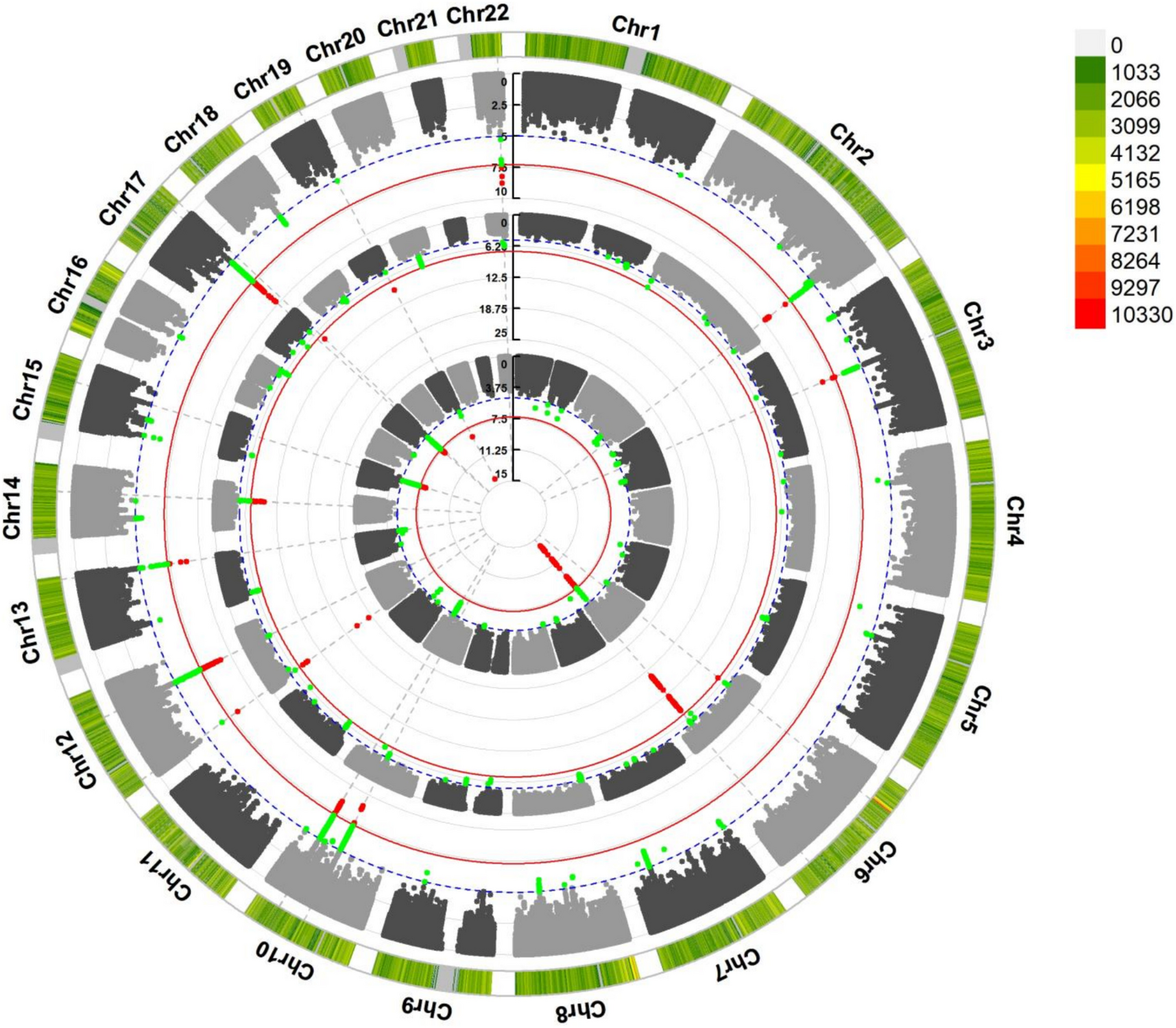
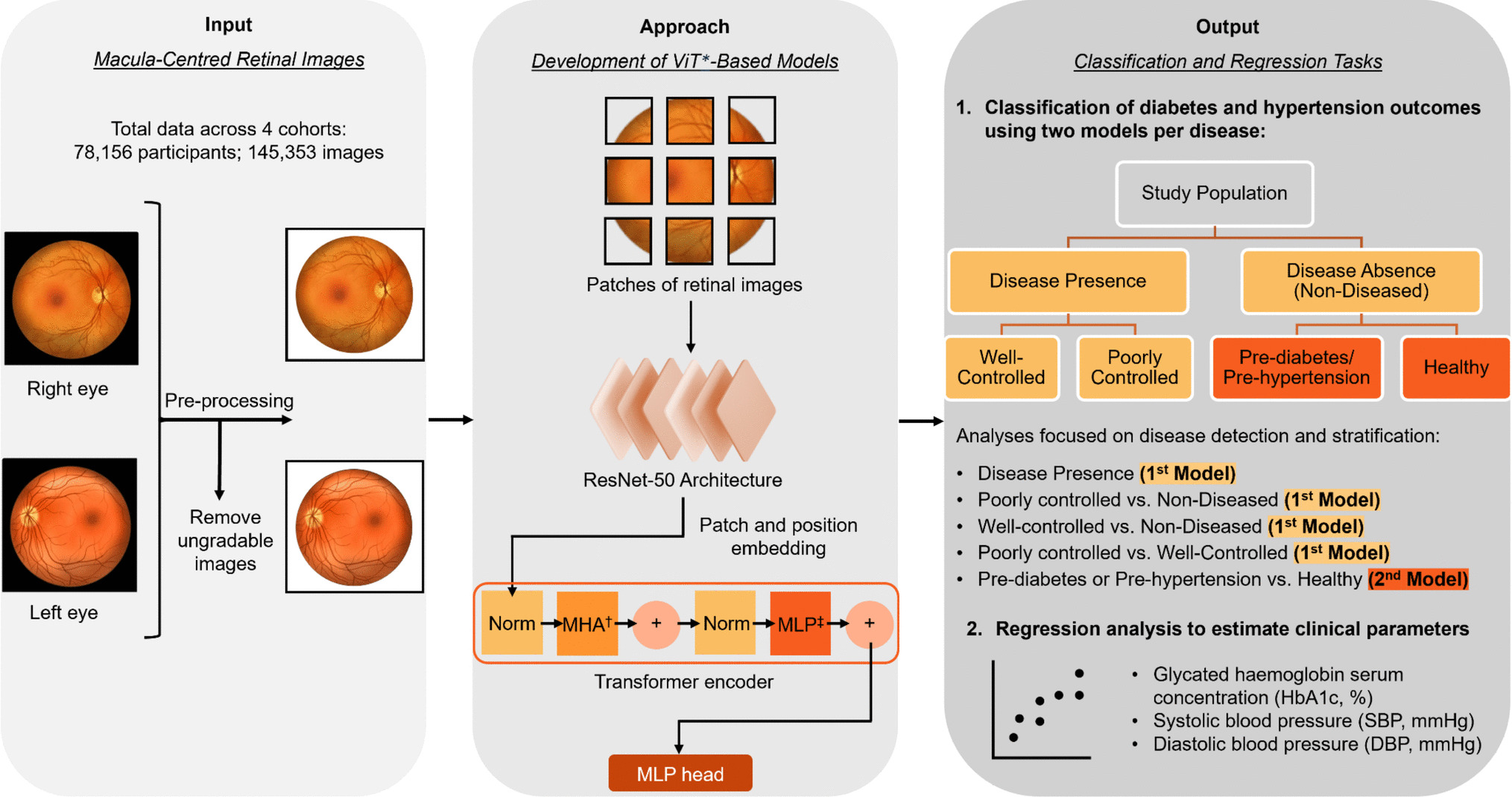
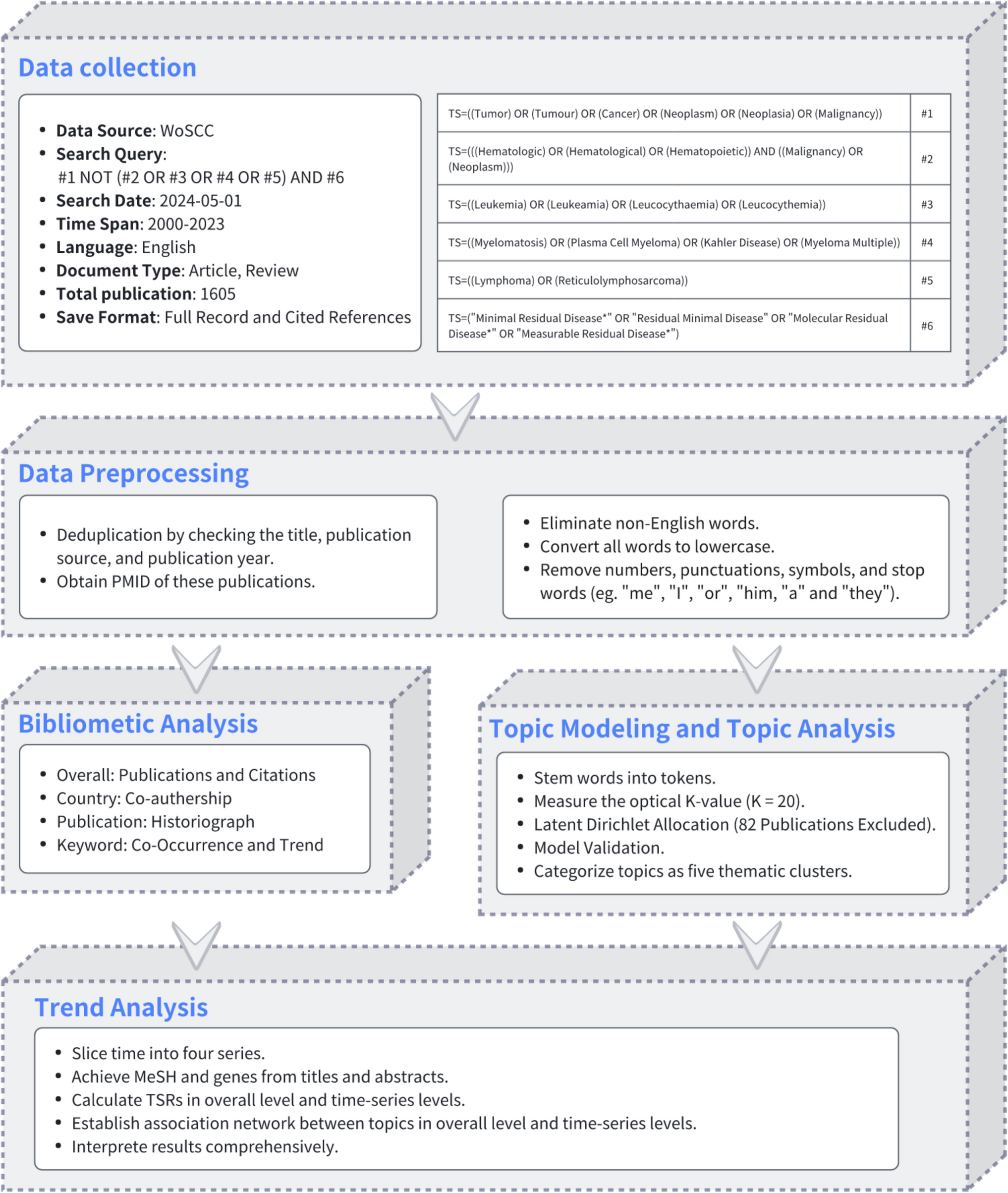

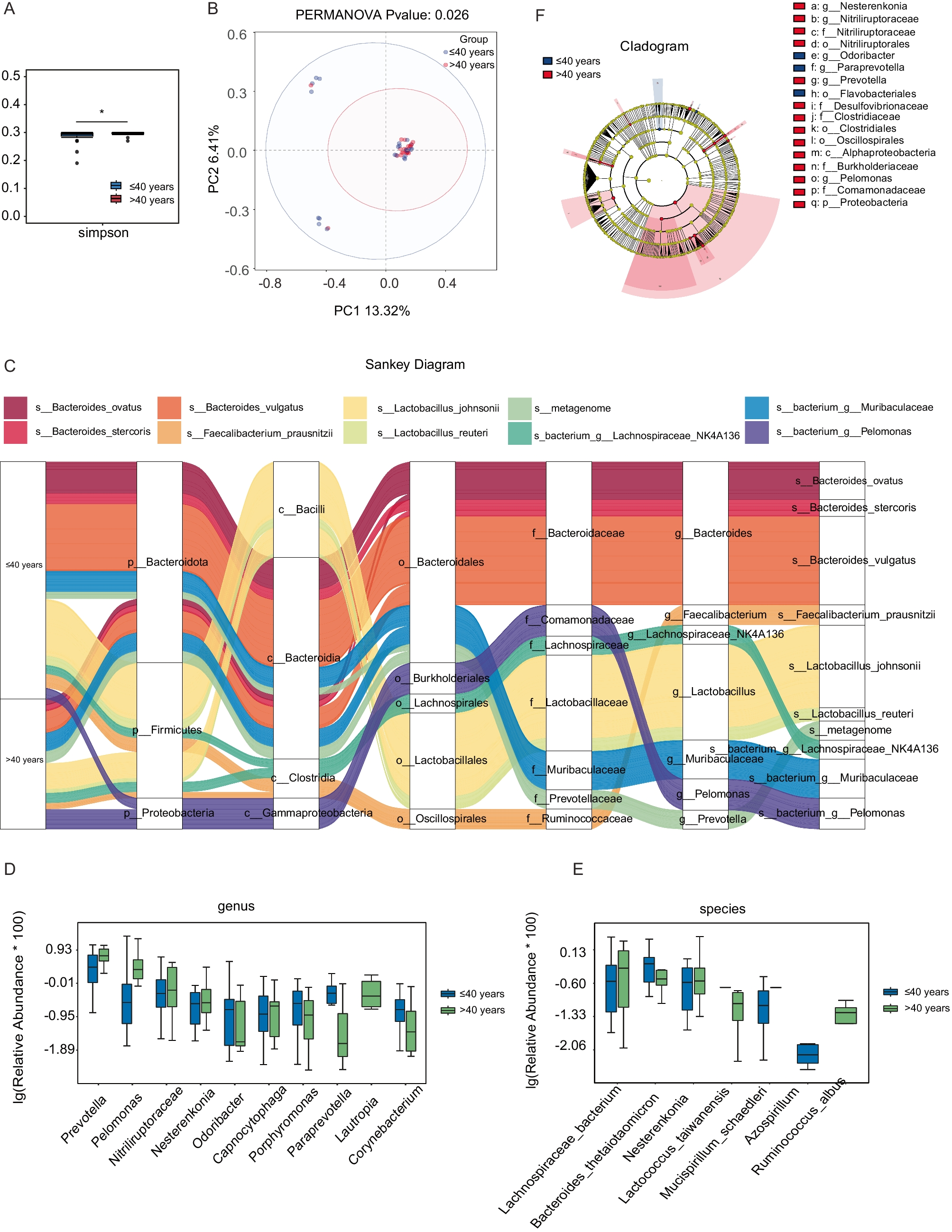


To investigate the potential influence of pelvic microbiota on ovarian reserve, ascitic fluid samples were collected from 45 patients undergoing surgery for benign gynecological tumors. Microbial composition and diversity were analyzed using 16S rRNA sequencing. Given the established association between age and ovarian reserve, patients were initially stratified into two age groups (≤ 40 years and > 40 years) to explore potential age-related shifts in microbiota that may contribute to diminished ovarian reserve. To assess overall microbial diversity, we conducted alpha diversity analysis, which measures species richness and evenness within each sample. The results showed that the Simpson index was significantly lower in the younger age group (age ≤ 40 years) P = 0.0344, indicating reduced microbial diversity (Fig. 1A). PCoA was then applied to visualize beta diversity, which evaluates differences in microbial community structure between samples. The PCoA plot, based on the Bray–Curtis distance, revealed distinct clustering between the two age groups, indicating significant microbial compositional differences (Permanova P = 0.026) (Fig. 1B). To further characterize the microbial composition of ascitic fluid, we constructed a Sankey diagram to visualize taxonomic distribution at different classification levels, from phylum to species (Fig. 1C). The dominant phyla across all samples included Bacteroidota, Firmicutes, and Proteobacteria. At the genus level, Bacteroides, Faecalibacterium, Lactobacillus, Muribaculaceae, Prevotella, and Pelomonas were predominant, with notable differences observed between the two groups. We further analyzed the differentially expressed microbial genera and species between the two groups (Fig. 1D, E). At the species level (Fig. 1E), Lachnospiraceae bacterium, Nesterenkonia, Lactococcus taiwanensis, and Ruminococcus albus were significantly overrepresented in the older age group. These findings suggest that specific microbial species may be associated with ovarian function alterations related to aging. To identify taxa with significant differential abundance between the groups, we performed linear discriminant analysis effect size (LEfSe) analysis (Fig. 1F). This method uses linear discriminant analysis (LDA) to determine the effect size of each taxon, allowing us to identify taxa that most strongly distinguish between the groups. The LEfSe results revealed several microbial genera and species that were significantly different between the younger and older age groups. Specifically, taxa such as Proteobacteria, Pelomonas, and Prevotella were significantly enriched in the older age group, with an LDA score greater than 104. These findings suggest potential microbial signatures that may be linked to ovarian function.
Fig. 1
Microbial composition and diversity of ascitic fluid stratified by age. A Alpha diversity analysis showed differences in microbial diversity between two groups, using the Simpson index (P = 0.0344 < 0.05, *). B PCoA revealed distinct clustering of microbial communities between the groups, based on Bray–Curtis distance (Permanova P = 0.026). C Sankey diagram illustrating the taxonomic distribution of microbiota from phylum to species level. D Differentially expressed microbial genera (top 10) were compared between the two groups using the Wilcoxon t test (P < 0.05). E Differentially expressed microbial species (top 10) were compared between the two groups using the Wilcoxon t test (P < 0.05). F LEfSe analysis identified microbial taxa significantly distinguishing the groups
This preliminary stratification highlights that age-related changes in the ascitic microbiome may reflect early shifts in ovarian function. However, chronological age alone may not fully capture the complexity of reproductive decline. Building on this observation, we propose that specific microbial signatures could serve as more sensitive and earlier indicators of DOR than conventional age or hormonal markers. Integrating these microbial profiles with clinical data holds potential for identifying high-risk individuals before symptoms appear, enabling timely lifestyle or probiotic interventions and promoting a shift toward proactive, personalized reproductive care.
Clinical and microbiota profiles in the study cohortIn the first part, we analyzed the ascitic microbiota based on age groups to assess general microbial differences across reproductive stages. To further clarify the association between pelvic microbiota and ovarian reserve, we then stratified patients by AMH levels. This approach facilitated the identification of DOR-related microbial features and enabled further clinical analysis of microbiome–ovarian function associations. The clinical characteristics of the study cohort are summarized in Table 1; a total of 45 participants were included, with 23 in the control group (AMH ≥ 1.1 ng/ml) and 22 in the DOR group (AMH < 1.1 ng/ml). Age was significantly higher in the DOR group (44.91 ± 6.63 years) compared to the control group (31.39 ± 5.92 years), with a P value of 0.000, indicating a strong association between age and ovarian reserve. The levels of FSH were also significantly elevated in the DOR group [12.95 (6.14–33.25) IU/l] compared to the control group [5.13 (3.74–6.63)] IU/l, with a P value of 0.000, suggesting a disrupted hormonal balance in the DOR group. Moreover, the gravity and parity of participants were significantly higher in the DOR group compared to the control group, with P values of 0.000. This increase may be attributed to the older age of participants in the DOR group, which is associated with a higher number of pregnancies and births. In terms of BMI (body mass index), the DOR group exhibited a significantly higher value [23.34 (21.42–24.71)] kg/m2 compared to the control group [20.58 (19.78–24.38)] kg/m2, with a P value of 0.038. This suggests that body weight may have an impact on ovarian function, with higher BMI potentially contributing to reduced ovarian reserve in the DOR group. Though no significant differences were observed between the two groups for several clinical parameters, including white blood cell (WBC), red blood cell (RBC), platelet (PLT), and CRP, certain immune-related markers, such as eosinophils (EOS) and basophils (BASO), exhibited trends toward differences that did not reach statistical significance. Furthermore, the prevalence of abnormal vaginal smear tests was higher in the DOR group (P = 0.057), suggesting a potential trend toward increased vaginal microbiota dysbiosis in individuals with diminished ovarian reserve.
Table 1 Characteristics of the study cohort. Data are presented as median (P25–P75) or mean ± SD, depending on data distribution. DOR group: AMH < 1.1 ng/ml (n = 22); control group: AMH ≥ 1.1 ng/ml (n = 23) (symbols represent significance levels: *P < 0.05; **P < 0.01; **P < 0.001)These findings suggest that clinical and hormonal markers, such as age, FSH, gravity, parity, and BMI, are strongly associated with ovarian function. Age-related ovarian decline is well-documented, with higher FSH indicating reduced reserve. Additionally, BMI may influence hormonal balance and metabolic status, indirectly impacting ovarian health. The observed associations between these clinical markers and pelvic microbiota patterns related to aging may offer predictive insights into the onset of diminished ovarian reserve. Integrating microbiota analysis with conventional clinical indicators may enhance early risk stratification of DOR. This combined approach supports targeted prevention strategies, such as lifestyle modification or probiotic interventions in high-risk individuals, thereby contributing to personalized and anticipatory reproductive care.
Following the comparison of baseline clinical characteristics between the DOR and control groups, we listed the differentially abundant bacterial genera in Table 2. Notable shifts were observed in both pathogenic and commensal taxa between the groups. Among the pathogenic genera, Capnocytophaga showed a higher median abundance in the DOR group [4.00 (IQR) 3.40–4.00)] compared to the control group [3.23 (IQR 2.63–4.00)]. In contrast, the genera Lautropia [4.00 (IQR 4.00–4.00)], Pelomonas [2.51 (IQR 1.94–3.07)], and Nesterenkonia [4.00 (IQR 3.47–4.00)] had higher median abundances in the control group compared to the DOR group [4.00 (IQR 2.68–4.00), 1.63 (IQR 1.31–2.01), and 2.62 (IQR 2.38–4.00)]. These findings suggest that Capnocytophaga may be associated with diminished ovarian reserve, while Lautropia, Pelomonas, and Nesterenkonia are more abundant in individuals with normal ovarian reserve, which may indicate that these potentially pathogenic genera are less likely to be involved in ovarian dysfunction.
Table 2 The differential distribution of genera in the DOR and control group. Bacterial abundance is shown as − log10 (X + 0.0001); values are presented as mean ± SD and median (IQR), depending on data distribution (symbols represent significance levels: *P < 0.05; **P < 0.01; **P < 0.001)Compared to the DOR group, the control group exhibited higher median abundances of UCG-002 [3.79 (IQR 2.58–4.00)] and Nitriliruptoraceae [4.00 (IQR 2.20–4.00)]. Conversely, the DOR group showed elevated levels of several other commensal genera, including UCG-005 [4.00 (IQR 3.88–4.00)], NK4A214_group [4.00 (IQR 3.69–4.00)], 0319-7L14 [4.00 (IQR 4.00–4.00)], Telmatospirillum [4.00 (IQR 4.00–4.00)], and Candidatus_Solibacter [4.00 (IQR 4.00–4.00)], all of which had lower median values in the control group. This shift in microbial composition suggests that the DOR-associated environment may favor the expansion of certain commensal taxa. While these bacteria are generally not considered pathogenic, their enrichment might represent a microbial response to underlying inflammation, hormonal dysregulation, or metabolic stress. Whether these changes serve a protective function or reflect early dysbiosis remains to be elucidated.
Interestingly, our findings suggest that diminished ovarian reserve is not necessarily associated with an increase in pathogenic bacteria. Instead, several commensal genera were more abundant in the DOR group. This unexpected shift in microbial composition may represent a compensatory or adaptive response to changes in the ovarian or pelvic microenvironment, rather than a consequence of overt microbial invasion. These results underscore the importance of further clinical and mechanistic investigations to clarify the functional role of commensal microbiota in ovarian health and their potential relevance in predictive modeling.
Association between altered microbiota and DOR riskThe relationship between the relative abundance of individual bacterial genera and the likelihood of DOR was assessed using multivariate logistic regression analysis, adjusting for relevant covariates. The results are visualized in Fig. 2. Among the taxa analyzed, high abundance of Capnocytophaga was significantly associated with an increased risk of DOR. Compared to individuals with low levels of Capnocytophaga, those with high levels had a markedly elevated likelihood of DOR (adjusted OR = 12.644; 95% CI, 1.399–114.279; P = 0.024). Although elevated levels of other potentially pathogenic bacteria, such as Lautropia, Pelomonas, and Nesterenkonia, also showed trends toward increased odds of DOR, these associations did not reach statistical significance (all P > 0.05). Similarly, the abundance of commensal taxa (UCG-002, UCG-005, 0319-7L14, Candidatus Solibacter) was not significantly correlated with DOR risk. Capnocytophaga was identified as a potential microbial biomarker associated with diminished ovarian reserve, warranting further investigation to clarify its mechanistic role and predictive relevance.
Fig. 2
Multivariate logistic regression analysis showing the association between the relative abundance of bacterial genera and the likelihood of DOR. ORs were adjusted for age, BMI, and other covariates. The vertical dashed line indicates OR = 1.0
Microbiota-based predictive modeling for ovarian reserveTo explore the potential of using pelvic microbiota and clinical parameters to predict the risk of DOR, we constructed multiple random forest classification models based on different feature sets, including eight key microbial genera, BMI, systemic inflammatory markers, and a combined model integrating microbial and metabolic features. The microbial genera were selected via multivariate logistic regression analysis (Fig. 2) based on their predictive contributions and biological plausibility. All models were trained using tenfold cross-validation to ensure robustness. Performance was assessed using ROC, with the AUC used as a measure of discriminative accuracy (Fig. 3).
Fig. 3
Predictive models for ovarian reserve risk based on microbial and clinical parameters: A model based on key microbial genera, B model based on BMI, C model based on inflammatory markers, D combined model of microbiota + BMI
The model based on key microbial genera showed moderate predictive power (AUC = 0.81 ± 0.22) (Fig. 3A), suggesting that certain taxa may be linked to the risk of DOR. This result indicates that microbial dysbiosis in the pelvic environment may serve as an early and specific signature for ovarian reserve estimation. In contrast, the BMI-only model exhibited moderate discriminative performance (AUC = 0.75 ± 0.22) (Fig. 3B), suggesting that metabolic status alone, as reflected by BMI, may be partially informative in predicting ovarian reserve. This finding is consistent with existing evidence that metabolic imbalance can affect reproductive hormones, follicular development, and ovarian aging [28]. The model based on systemic inflammatory indicators (CRP and complete blood count) showed the lowest predictive value (AUC = 0.58 ± 0.29) (Fig. 3C), highlighting that systemic inflammatory markers have limited predictive value for ovarian function, as diminished ovarian reserve appears to be more closely associated with localized pelvic microenvironmental shifts and chronic metabolic dysregulation rather than with overt systemic inflammation. To improve predictive sensitivity, we developed an integrated model combining microbial features and BMI, which achieved the highest performance (AUC = 0.88 ± 0.16) (Fig. 3D), offering a more comprehensive risk stratification approach for early detection of DOR.
These findings demonstrate that the integrated model, which combines pelvic microbial signatures with BMI, serves as an effective tool for both prediction and early diagnosis of diminished ovarian reserve, while also supporting individualized risk stratification through the inclusion of the metabolic indicator: BMI. Given the importance of translating early diagnostic insights into actionable preventive strategies [29], we next investigated the function of the identified microbial genera to explore their potential roles in ovarian health preservation.
Functional potential of ascitic microbiotaWith the correlation analysis results above, we found that pelvic microbiota dysbiosis is indeed a key factor influencing ovarian function. To investigate the functional potential of the ascitic microbiota, we first performed a Kyoto Encyclopedia of Genes and Genomes (KEGG) functional classification analysis to examine the microbial community’s overall functional profile. This analysis revealed a diverse array of pathways related to membrane transport, cellular community (prokaryotes), metabolism of other amino acids, xenobiotics biodegradation and metabolism, cell motility, infectious disease (bacterial), and cancer (specific types), indicating a potential impact of the microbiota on ovarian function (Fig. 4A). To further investigate the correlation between KEGG pathways and ovarian function, we analyzed the differentially enriched pathways the preserved ovarian function group and the DOR group (using AMH 1.1 ng/ml as the threshold, with control group representing AMH ≥ 1.1 ng/ml and DOR group representing AMH < 1.1 ng/ml). Following this, we found several KEGG pathways that differed significantly between the two groups. Specifically, microbial metabolism in diverse environments, adenosine triphosphate (ATP)-binding cassette (ABC) transporters, biosynthesis of nucleotide sugars, and quorum sensing were significantly enriched in the DOR group (Fig. 4B, C). These pathways may influence ovarian function and the aging process by regulating ovarian energy metabolism, DNA/RNA synthesis, cell proliferation, protein synthesis, and microenvironment modulation. We further analyzed the correlation between these differentially expressed pathways and ovarian function markers: AMH, LH, FSH, and E2. The results revealed that amino sugar and nucleotide sugar metabolism and nucleotide metabolism were positively correlated with E2, suggesting a potential role in estrogen biosynthesis, follicular development, and ovarian cellular function maintenance (Fig. 4D). These findings indicate that the identified microbial genera are not only useful for prediction and diagnosis but may also influence ovarian function through specific metabolic and endocrine-related pathways. Their potential involvement in hormone regulation and metabolic balance suggests that changes in the pelvic microbiota could contribute to diminished ovarian reserve. This highlights the value of developing preventive strategies that target microbiota-related dysregulation and supports the integration of microbial features into personalized clinical interventions.
Fig. 4
Functional potential of ascitic microbiota associated with DOR. A KEGG functional classification analysis of ascitic microbiota. B Differentially expressed KEGG pathways between the groups. C Top 6 differentially abundant KEGG pathways between the groups. D Correlation analysis between differential KEGG pathways and ovarian function markers (AMH, LH, FSH, E2) (control group: non-DOR group, AMH ≥ 1.1 ng/ml, DOR group representing AMH < 1.1 ng/ml) (correlation method: Spearman. Symbols represent significance levels: *P < 0.05, **P < 0.01, **P < 0.001, ***P < 0.0001)
Comments (0)