
Remember me


Tables 2, 3 and 4 summarize the results of the ADM analysis for each protein in this study, along with statistics on conserved hydrophobic residues and the detailed positions of F-value plot peaks.
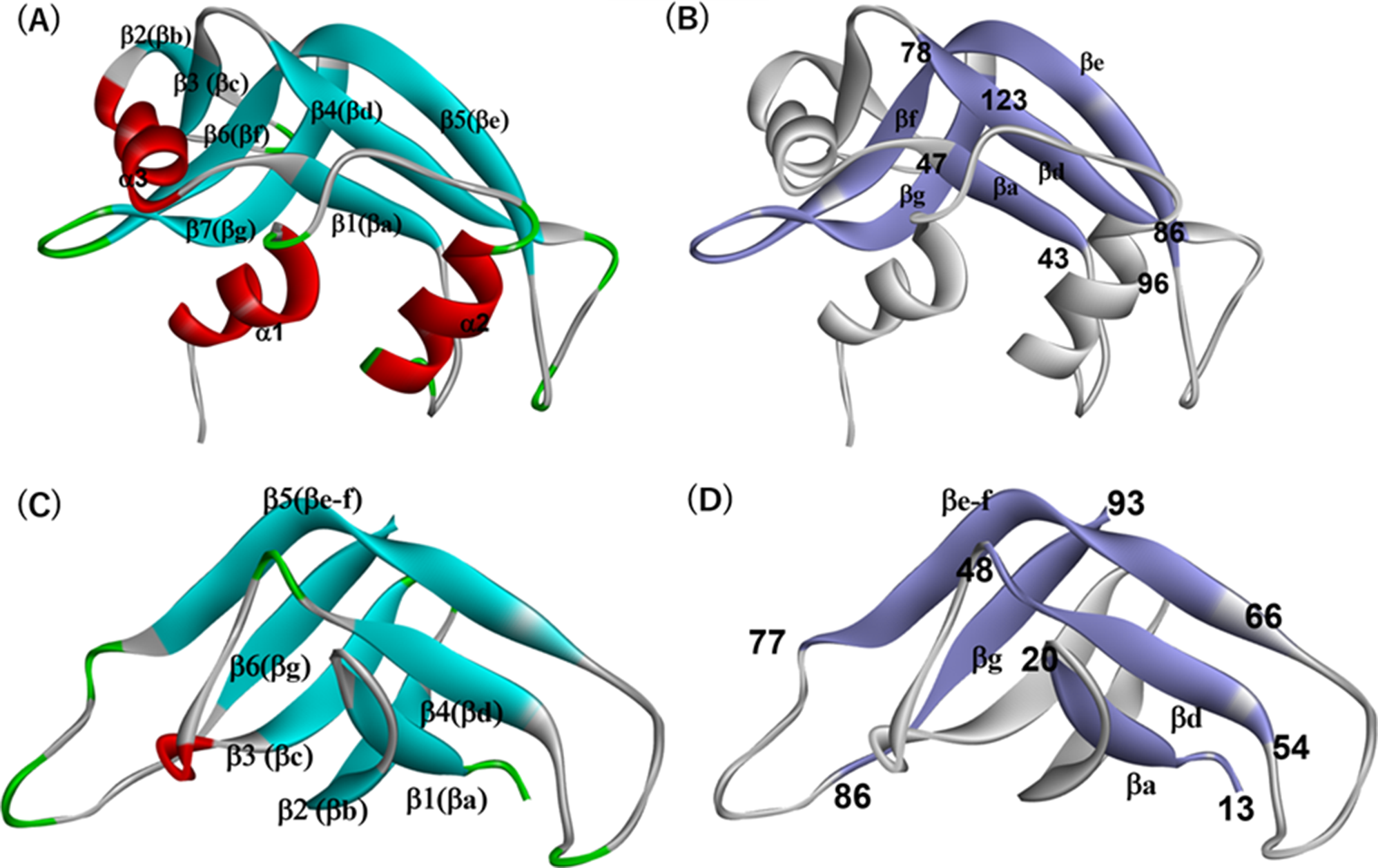
Table 2 Summary of the ADM predicted regions in every study proteinTable 3 Statistics of conserved hydrophobic residues in study proteinsTable 4 The summary of the F-value peak positionsRNase A-like foldThe location of the corresponding common segments in ribonuclease A (6ETL) is illustrated in Fig. 1(B). Figures 4 and 5 present the ADM and F-value analyses for 6ETL, respectively, alongside data from the NMR H/D exchange experiment by Neira et al. [39]. The ADM analysis predicts two predicted compact regions (PdCR), the N-terminal region from residue 19–84 is the prominent structured unit compared to their C-terminal region, which is from the residue 92 to 118 and their h-values (strength of compactness), 0.219 and 0.146 respectively. In Fig. 4(B), these ADM-predicted regions are highlighted in red and green in the 3D structures of 6ETL (see also Table 2), with the higher η-value region colored red (From here, the red and green colors are used to distinguish PdCRs).
Fig. 4
(A) ADM for 6ETL. The location of a secondary structure is indicated by a bar or an arrow for a-helix or b-strand along the diagonal. The red (green) triangle denotes PdCR with a higher (lower) h value. (B) The regions of PdCRs in the 3D structure of 6ETL. The red (green) part denotes PdCR with a higher (lower) h value (See Table 2 for the assignment of the second PdCR)
Fig. 5
F-value plot with the NMR H/D exchange experimental data [39] for 6ETL. The blue line denotes the F-value plot. A residue number indicates the position of a peak. The H/D exchange free energy of a residue is characterised by an orange bar. The value of exchange free energy for a residue was estimated from the experimentally obtained H/D exchange rate constant of a corresponding residue [39]. The value of exchange free energy for a residue is sometimes presented as > 9.5 and so on in [39]. The number of the orange lines with the broken lines is 19. To explain this situation, we use an orange broken line for the corresponding values. A red dot denotes the position of a CHR. The x-axis denotes the residue number. A grey bar and a grey arrow on the x-axis mean a position of a-helix and b-strand, respectively. In the lowest part of this figure, two PdCRs are indicated by the red and green bars
The first PdCR (residues 19–84) includes the β1 and β4 strands from the common segments, while the second PdCR (residues 92–118) corresponds to β5, β6, and β7. The interaction between these two PdCRs is likely to contribute to the formation of the common structure.
Taking higher h value into account, the N-terminal site is expected to form an initially stable folding core during the folding. The F-value plot of 6ETL has six high peaks as presented in Table 4. The peaks are in α2, α3, β2, β3, and β6. The first PdCR includes the α2-α3 and β1-β4; on the other hand, the second PdCR includes the β5, β6 and a part of β7. The F-value analysis in Fig. 5 indicates that the α3 and β6 are the keys to their folding because they have high contact frequency. NMR H/D exchange result showed that the highly protected parts are the α3 and β6, as shown in Fig. 5. The orange bars with broken lines in Fig. 5 correspond to residues with high H/D exchange free energy from the study by Neira et al. [39] (see also the legend of Fig. 5 in detail). The number of such residues is 19. Among these 19 residues with high H/D exchange free energy, 12 residues are near the peaks of the F-value plot within ± 4 residues, with 11 of these residues clustered around the two highest peaks near conserved hydrophobic residues (CHRs, red dots in Fig. 5), namely 57-Val and 108-Val. This suggests that these residues are likely centers of folding during the early stages. Notably, α3 and β6, which are part of the first and second PdCRs, respectively, act as folding centers, with α3 serving as the leading folding initiation site due to its higher η-value. The number of these residues with high H/D exchange free energy are 19 and, among them, 8 residues are within the first PdCR and 8 residues are within the second PdCR, that is, two folding initiation sites identified by H/D exchange experiment are in respective PdCRs. The h value of the second one is smaller than the first one, and the peak of the F-value plot around 108-Val is relatively low. Therefore, the main center is the first ADM-predicted region, and the interaction with the first one may stabilize the second one.
In the RNase ZF-3E (PDB code: 2VQ9) protein, the corresponding common segments β1, β4, and β6 are presented in Fig. S1(B). Notably, the corresponding β5 and β6 strands in 6ETL are combined into β5 in 2VQ9 according to PDB annotations. ADM analysis of 2VQ9 reveals a highly compact N-terminal region (residues 8–85) with an η-value of 0.227, encompassing α1-α3 helices and β1-β4 (Fig. S4(A), (B)). The second PdCR (residues 93–109) corresponds to β5 with an η-value of 0.108. Similar to 6ETL, the first PdCR in 2VQ9 exhibits higher compactness, suggesting that it forms a stable folding core, while the second PdCR acts as a smaller structural unit stabilized by the interaction with the first PdCR. Fig. S4(C) shows the peaks of the F-value plot for 2VQ9, which are located near conserved hydrophobic residues. The two highest peaks (positions 56 and 80) in α3 and β4 are likely early-stage folding sites, forming part of the first PdCR. Additionally, a high F-value peak at 109-Cys in β5 indicates another compact predicted region in the C-terminal region, suggesting that the N-terminal region initiates folding, similar to 6ETL. As no experimental data on the folding of 2VQ9 is available, comparisons can only be made with the experimental data for 6ETL.
The ADM analysis of turtle egg white ribonuclease (PDB code: 2ZPO) shows a similar pattern to the other RNase A-like fold proteins, with two PdCRs identified (Fig. S5(A), (B)). The primary PdCR spans residues 1–81 with an η-value of 0.372, and the secondary PdCR spans residues 92–107 with an η-value of 0.154. The first PdCR is predicted to have a higher potential for initiating folding compared to the second PdCR. The F-value plot of 2ZPO, presented in Fig. S5(C), has three peak positions at the residues in Table 4. Three peak positions in α3, β4, and β6 indicate that these regions are critical to folding. The early-stage folding likely begins with the helix in the region 1–81 which then forms a core folding unit with the β6 strand, stabilized by residues 92–107.
Interestingly, despite the relatively low sequence identity among these RNase A-like fold proteins (25–40%), they all exhibit similar PdCRs according to ADM analyses. Each protein has two predicted compact regions, with the N-terminal region being more compact than the C-terminal region (Table 2). Importantly, the second PdCR in each protein contains β5 and β6 strands, which are part of the common 3D topology described in Figs. 1 and S1. The NMR H/D exchange experiment for 6ETL showed that the α3 helix, β3, and β6 strands exhibit the slowest exchange rates (high H/D protection), forming a highly stable hydrophobic core [39,40,41]. ADM analysis predicts two PdCRs, which include the α3 helix and β3 strand in the primary compact region and the β6 strand in the secondary compact region. Based on these findings, we can infer that the first PdCR is likely the folding initiation site for all these proteins, with the interaction between the PdCRs forming a stable folding core during the folding process, consistent with experimental results [39].
Trypsin-like serine proteases foldADM prediction for 6CHA domain B exhibits two compact regions. One is from residue 26 to 53, and the other is from residue 65 to 128, illustrated in Fig. 6(A). The highly compact region in the N-terminal has the h-value of 0.188 (first PdCR) that includes α1 helix and β2-β4 (bb-bd). Another one has the h-value of 0.180 (second PdCR), which contain β5 (be-f) and β6 (bg) (as mentioned b5 (be-f) in 6CHA corresponds to b5 (be) and b6 (bf) in 6ETL, that is, one b strand in the PDB annotation). The compactness of the predicted regions is quite similar, and their N-terminal region is slightly higher than the second PdCR. As shown in this figure, the N-terminal compact region covers β2(bb), β3(bc), α1 and β4(bd) secondary structures. The F-value plot is in Fig. 7. The highest F-value peak appears at β3 at residue number 38-Val. The second PdCR ranges from β5(be-f) to b7 and has four high peak positions as Table 4 presents. These are possible folding sites. From the results of the F-value analysis, we can expect that the β3(bc), b5(be-f), and β6(bg) strands will make the folding core in the common topology. The meaning of the peaks at 106-Val and 120-Thr will be discussed later.
Fig. 6
(A) ADM for 6CHA (For the construction of ADM, the whole sequence of 6CHA is used). The location of a secondary structure is indicated by a bar or an arrow for a-helix or b-strand along the diagonal. The red (green) triangle denotes PdCR with a higher (lower) h value. (B) The regions of PdCRs in the 3D structure of 6CHA (Only the structure of the core part (not the whole sequence) is shown). The red (green) part denotes PdCR with a higher (lower) h value
Fig. 7
F-value plot for 6CHA. The blue line denotes the F-value plot. An arrow with the corresponding residue indicates the position of a peak. A red dot represents the position of a CHR. At the bottom of the figure, the x-axis denotes the residue number. A grey bar and a grey arrow on the x-axis mean a position of a-helix and b-strand, respectively. In the lowest part of this figure, two PdCRs are indicated by the red and green bars
Though the results of our present analyses should be examined by objective experimental data, to our best knowledge, no experimental analyses on folding of a protein in Trypsin-like serine proteases fold have been performed so far.
Figure S6(A), (B) and Table 2 represent the ADM of 1LTO having two PdCRs, the first PdCR includes residues 12–60 with η value 0.164, and the second PdCR includes residues 65–112 (η value of 0.277). The C-terminal site contains α3 helix and β6-β7strands, which are expected to be stable in the early stage of folding based on the h values. The F-value for 1LTO shown in Figure S6 (C) indicates that there are three prominent peaks at 38-Trp (in β4), 78-Ile (in b6) and 103-Ile (near b7) (see also Table 4), predicting that the β4 and β7 strands with many CHRs are significant for their folding of this protein. The hydrophobic residues in β4 and β7 are highly conserved during the evolution, that is, a lot of CHRs in β4 and β7. Thus, β4 and β7 strands seem to be very important for the folding of this protein. Those high peaks at β4 and β7 are in two different PDCRs. It is plausible that these conserved hydrophobic residues near an F-value peak are significant in stabilizing the interaction between two PdCRs. From here, we use the abbreviation “CHRnF” as conserved hydrophobic residues near an F-value peak.
ADM and F-value results of 3DFJ are shown in Figs. S7 (A)-(C) (See also Table 2). Two PdCRs are predicted by ADM; the first PdCR covers 7–43 with a 0.232 h value, and the second one is 76–114 with h value of 0.251. F-value analysis in Fig. S7 (C) exhibits six peaks in the data plot, and many CHRnFs are distributed around the peak at 26-Cys and 36-Val in the first PdCR and 93-Leu in the second PdCR (Table 4). Thus, these CHRnFs would be prominent for the folding of 3DFJ. Considering these results, we can assume that the folding core will be made by β3-β4 and β8 strands.
In conclusion, our analysis of ADM predictions, F-value data, and conserved hydrophobic packing suggests that the folding properties of all studied proteins within the Trypsin-like serine proteases fold are remarkably consistent. These findings highlight the critical role of conserved hydrophobic residues and their interactions in guiding the folding process and stabilizing the protein structure.
Evolutionary analysis of the RNase A-like fold and trypsin-like serine proteases fold proteinsWe examined the evolutionary conservation of predicted folding units (PdCRs) in RNase A-like and trypsin-like serine proteases fold proteins using multiple sequence alignments combined with ADM results for homologous sequences obtained via BLAST search. Figs S8(A)-(F) show these alignments, with PdCRs highlighted in red. A histogram below each figure, indicated by a blue line, shows the ratio of residues at aligned sites within PdCRs to the total number of aligned sequences. Higher ratios suggest that these regions are evolutionarily conserved folding units. Table 1 represents the sequence identities of around 33–40% within the same group but only about 10% identity between proteins from different groups.
In ribonuclease A (6ETL), the alignment of 76 sequences (average sequence identity ~ 38%) shows that regions with higher η-values are concentrated in the N-terminal region illustrated in Fig. S8(A). Conserved folding units include β1, α3, β2, β4, and regions containing β5 and β6. All 11 conserved hydrophobic residues are within PdCRs, with 7 near peaks in the F-value plot (Table 5). It is important to note that the properties of PdCRs and F-value profiles exhibit considerable variability among the proteins analyzed here. Despite these differences, the overarching features, such as critical folding nuclei, remain consistent across homologous proteins. This variability may reflect nuances in the specific folding properties of homologous proteins, as discussed in references [18–19], and [22].
Table 5 The summary of the sequence for the evolutionary analysesThe plot denoted by an orange line in Fig. S8 indicates a ratio of hydrophobic residues to the total number of sequences at an aligned site. We regard a more than 90% ratio that the current site shows the conservation of hydrophobic residues. The conserved hydrophobic residues in PdCRs are indicated by yellow letters, and out of PdCRs are indicated by blue letters in this figure. For 6ETL, the conservation of hydrophobic residues is observed at 11 sites in Fig. S8(A), and all of the conserved hydrophobic residues are in PdCRs of 6ETL. If we consider a region with more than 70% ratio of conservation of predicted regions tentatively (indicated by an orange line in the bottom of Fig. S8(A)) as an evolutionarily conserved predicted unit, then 11 out of the 11 conserved hydrophobic residues are included in the evolutionarily conserved predicted unit (The blue line above the orange line in the bottom of Fig. S8(A)). Moreover, 7 of these residues are near the peaks of the F-value plot of 6ETL within ± 5 residues represented in Fig. 5; Tables 6 and 7.
Table 6 Table of conserved hydrophobic residuesTable 7 Conserved hydrophobic residues near F-Value peak (CHRnFs)For RNase ZF-3E (2VQ9), analysis of 78 sequences (average sequence identity ~ 40%) reveals that PdCRs include β1, β2, β4, α3, and β5-β6 (Fig. S8(B)). The histogram indicates that over 70% of conserved predicted folding units involve these regions, which contain 9 out of 10 conserved hydrophobic residues, 6 of which are near F-value peaks (Fig. S4(C), Tables 3, 6 and 7).
In turtle egg white ribonuclease (2ZPO), analysis of 29 sequences (average sequence identity ~ 33%) shows that high η-values are primarily in the N-terminal region, with PdCRs covering α2-β4 and β5-β6 as shown in Fig. S8(C). More than 70% of conserved folding units involve β1-β3 and β4-β6, with 6 out of 11 CHRnFs located within these regions (Fig. S5(C), Table 7).
The sequence alignment of α-chymotrypsin domain B (6CHA) is shown in Fig. S8(D), incorporating 110 sequences with an average sequence identity of about 31%. The alignment results suggest that both N-terminal and C-terminal regions exhibit similar levels of conservation, covering β1-β4 and β5-β7, respectively. Highly conserved regions are observed in β2, β3, α1, β4, β6, and β7, with more than 70% conservation. Of the 22 conserved hydrophobic residues identified, 18 are located within these highly conserved regions, all of which correspond to peaks in the F-value plot (Fig. 7; Tables 6 and 7).
Figure S8(E) shows the multiple sequence alignment for α1-tryptase (1LTO), which included 117 sequences with an average sequence identity of approximately 32% (Table 5). ADM analysis predicts two folding units: one in the N-terminal region covering β3 to α2, and another in the C-terminal region covering β6 to β7. Of the 18 conserved hydrophobic residues, 14 are located within these evolutionarily conserved units, and 9 are near F-value peaks within ± 5 residues (Fig. S6(C), Tables 6 and 7).
Finally, the analysis of prostasin (3DFJ) shown in Fig. S8(F) includes 76 sequences with an average sequence identity of 31%, with maximum and minimum identities of 89% and 11%, respectively. The regions with higher η-values are primarily located in the C-terminal region. The conservation of hydrophobic residues is observed at 16 sites, all within PdCRs. The histogram indicates that β4, α1, β6, and β8 are evolutionarily conserved folding sites, with 13 out of 16 conserved hydrophobic residues located within these regions, adjacent to F-value peaks (Fig. S7(C), Tables 6 and 7).
Hydrophobic packing of evolutionarily conserved residuesIn this section, we analyze the interactions of conserved hydrophobic residues within predicted folding units (PdCRs) based of the 3D structures of the studied proteins. The basic assumption is that conserved hydrophobic residues near F-value peaks (CHRnFs) are buried early in protein folding, serving as folding initiation residues [23–24, 26]. The folding process is thought to progress mainly through interactions among CHRnFs and other conserved hydrophobic residues (CHRs).
We start by identifying CHRnFs near the highest F-value peak within \(\:\pm\:\)5 residues [26] for each protein and then examine the compact regions (PdCRs) where these residues are located. We investigate the hydrophobic contacts within these PdCRs and between them, summarizing the results as contact maps (e.g., Fig. 8 for 6ETL). In the main text, we focus on interactions between secondary structure elements containing CHRnFs or CHRs, with detailed descriptions provided in the figure legends.
Fig. 8
Kind of contact map for 6ETL. Conserved hydrophobic residues are mainly presented. A red or green colored residue means a concerned residue is in a PdCR with the same color as in Fig. 4. The secondary structure containing a concerned residue is also indicated. A residue with an underline denotes CHRnF. A residue within a parenthesis is not CHR nor CHRnF. A contact colored by yellow means stabilization of a part of the common structure and a contact colored by violet means a contact between a residue in the common structure and a3. The CHRnFs in the second PdCR, 92–118, that is, 106-Ile and 108-Val make hydrophobic packing with 120-Phe
RNase A-like fold proteinsFor ribonuclease A (6ETL), the PdCRs are located at residues 19–84 and 92–118, with the first PdCR exhibiting a higher η-value (Table 2). Figure 5 indicates that the highest F-value peak, 57-Val in α3, is a CHRnF within the first PdCR, suggesting α3 as the folding center. Figure 8 shows that hydrophobic contacts form among CHRnFs and CHRs in α2, α3, β1 (βa), and β4 (βd) within this region. The second PdCR, spanning residues 92–118, has a peak of the F-value plot at 108-Val as shown in Fig. 5 (a CHRnF in β6). Contacts between α2 and β6, as well as among α3, β4, β5 (βe), and β6 (βf), indicate interactions between the first and second PdCRs, stabilizing the common structure (Figs. 8 and 9(A)-(E)). The α3 helix can be regarded as a center of contacts that support the formation of the common structure. In Fig. 8, contacts colored yellow indicate those that directly stabilize the common structure, while contacts colored violet signify those that provide stabilization through their interaction with α3.
Fig. 9
(A) Hydrophobic packing formed by CHRnFs (indicated by a bold stick representation), 54-Val and 57-Val, and CHRs (indicated by a thin stick representation), 47-Val and 79-Met stabilizing the interactions among a3, b1 and b4 within the first PdCR in 6ETL. (B) Hydrophobic packing formed by CHRnFs, 29-Met and 30-Met, and CHR, 46-Phe stabilizing the interactions between a2 and b1 within the first PdCR. The interaction between CHRs, 47-Val and 81-Ile, stabilizes interaction between b1 and b4 within the first PdCR. (C) Hydrophobic packing formed by CHRnFs, 54-Val and 57-Val, 106-Ile and 108-Val stabilizing the interactions between a3 and b6, that is, the interaction between the first and second PdCRs. (D) 102-Ala makes a contact with 81-Ile and this contact contributes to the stabilization of the b-sheet between b4 and b5. 102-Ala is relatively close to a peak of the F-value plot at 108-Val (Fig. 5) separating by 6 residues. 102-Ala is not CHR but expected to be involved in the early stage of folding though it may be not so strong. (E) Hydrophobic packing is formed by CHRnFs, 106-Ile,108-Val and a hydrophobic residue 120-Phe within the second PdCR. 120-Phe, a residue which is not CHR nor CHRnF, is indicated by a line representation. The corresponding residue appears in both 2VQ9 and 2ZPO (See Figs. S9, S10, S11 and S12). It is considered that the 3D structure of the region 92–118 is stabilized mainly by the interaction with the region 19–84. That is, PdCR 92–118 is the case in Fig. 3(E); that is, the region 92–118 interacts with the major part of the other part from this region (see the legend of Fig. 9(E)). See the legend of Fig. 3(E)
For RNase ZF-3E (2VQ9), PdCRs are at residues 8–85 and 93–109 (Table 2), with 56-Thr in α3 being the highest F-value peak, and CHRnF near to this residue is 57-Val (Fig. S4). Similar to 6ETL, α3 is predicted as the folding center, with hydrophobic contacts forming among CHRs in α2, α3, β1 (βa), and β4 (βd) within the first PdCR (Figs. S9, S10(A), (B)) stabilizing a part of the common structure (Fig. S9). The second PdCR, with a peak at 109-Cys (the CHRnF is 107-Ile in β6(bf). See Fig. S4(C).), also shows interactions between β6(bf) and β7(bg) within this PdCR. Furthermore, a contact between b1 and β6, that is, the interactions between PdCRs is also observed (Fig. S9). The contacts among a2, a3, b4(bd), b5(be) and b6(bf) also stabilizing the interaction between two PdCRs and leading to stabilize the common structure under the support of a3 (Figs. S9, S10(C)-(D)).
In turtle egg white ribonuclease (2ZPO), PdCRs are located at residues 1–81 and 92–107 (Table 2), with 55-Thr in α3 identified as the highest F-value peak. CHRnF, 54-Ile, in α3, regarded as a folding center in 2ZPO, forms contacts with β1 (βa) and β4 (βd), stabilizing the structure within the first PdCR with higher h-value (Figs. S11, S12(A)-(B)). Fig. S11 also indicates the contacts among a2, a3, b1(ba) and b4(bd) within the first PdCR stabilizing a part of the common structure. The second PdCR, with an F-value peak at 103-Ile (CHRnF) in β6(bf), shows interactions between β6(bf) and β7(bg), further stabilizing the structure (Figs. S12C-D). Interactions among β1(ba), β4(bd), β6(bf) directly stabilize the common structure, and α3, similar to 6ETL and 2VQ9, reinforces the common structure (Figs. S11 and 12(A), (C)).
Trypsin-like serine proteases foldIn α-chymotrypsin domain B (6CHA), PdCRs are located at residues 26–53 and 65–128, with a slightly higher η-value in the first PdCR (Table 2). As presented in Fig. 7, the highest F-value peak at 38-Val (a CHRnF in β3) suggests that β3(bc) is the folding center. Hydrophobic contacts among β2(bb), β3(bc), and β4(bd) stabilize the four stranded partial β-barrel structure (Figs. 10 and 11(A)), with additional interactions between β5(be-f) and β6(bg) contributing to the stability of the second PdCR (CHRnF is 90-Ile in b5(be-f) in this PdCR. Figures 10 and 11(A)-(C)). The common structure is stabilized by direct interactions among β1 (βa), β4 (βd), β5 (βe-f), and β6 (βg), as indicated by contacts colored yellow in Fig. 10. Additionally, β3 (βc) plays a supportive role in these interactions, as reflected in the violet-colored contacts in Fig. 10 (see also Fig. 11(D)).
Fig. 10
Kind of contact map for 6CHA. Mainly conserved hydrophobic residues are presented. A red or green colored residue means a concerned residue is in a PdCR with the same color as in Fig. 6. The secondary structure containing a concerned residue is also indicated. A residue with an underline denotes CHRnF. A residue within a parenthesis is not CHR nor CHRnf. A contact colored by yellow means stabilization of a part of the common structure and a contact colored by violet means a contact between a residue in the common structure and b3(bc)
Fig. 11
(A) Hydrophobic packing formed by CHRnFs (indicated by a stick representation), 36-Trp and 38-Val, and CHR (indicated by a line representation), 32-Ile stabilizing the interactions between b2 and b3 within the first PdCR in 6CHA. (B) Hydrophobic packing formed by CHRnF, 37-Val, and CHR, 31-Leu stabilizing the interactions between b2 and b3 within the first PdCR. 31-Leu and 37-Val are also interacting hydrophobic residue, 14-Trp, 16-Val and 18-Leu stabilizing the interactions among b1, b2 and b3 within the first PdCR. (C) Hydrophobic packing formed by CHRnFs, 74-Phe and 90-Leu stabilizing the interactions between b5 and b6 within the second PdCR. (D) Hydrophobic packing formed by CHRnFs, 36-Trp, 74-Phe and 90-Leu stabilizing the interaction between the first and second PdCRs. (E) Hydrophobic packing formed by CHRnF, 91-Leu and 18-Leu stabilizing the interaction between b1 and b6. (F) Hydrophobic packing is formed by CHRnF, 37-Val, and hydrophobic residue, 51-Val stabilizing the interaction between b1 and b3. There is no CHRs in b4, and b4 seems not to be actively involved in the folding of the first PdCR. However, CHRnF, 37-Val makes a contact with 51-Val in b4. This contact stabilizes the interaction between b3(bc) and b4(bd) although this contact would not be so strong during the folding. Furthermore, CHRs, 16-Val and 18-Leu make hydrophobic contact with 51-Val indicating the stabilization of b1(ba) and b4(bd), that is, a part of the common structure
106-Val and 122-Val are CHRnFs (see Table
Comments (0)