This research demonstrates how the usage of longitudinal omics data along with neuro-axonal damage marker can pinpoint the pathways and genes through which TMT induces neurotoxicity and neuro-axonal damage and links that to various neurodegenerative conditions, including Alzheimer's disease.
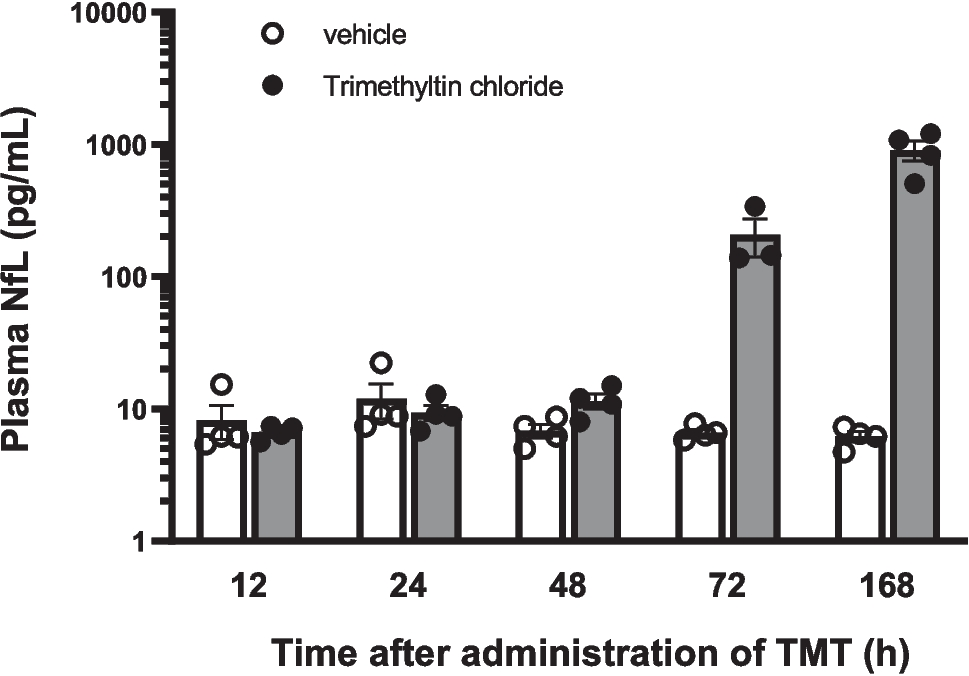
The regulation of genes/proteins induced by TMT appears to happen gradually, with the peak occurring at 72 h and 168 h following administration (Fig. 2), which marks the time of neuro-axonal damage indicated by elevated NfL levels (Fig. 1). This gradual increase in response may be attributed to a dose-time dependent relationship, where the maximum response is observed during the later stages (Subashika et al. 2022) Consequently, it is crucial to measure gene/protein expression both in the early and late stages of neurotoxicant administration. Additionally, it is possible that certain transcription factors were activated during the early stages and required time to exert their influence on the expression of other genes. Alternatively, the neuro-axonal damage in some cells indicated by elevated NfL levels might have triggered differential gene/protein expression in other cells as well, increasing the number of differentially expressed genes and proteins at 72 h and 168 h after TMT administration.
In the TNF signaling pathway, several genes that contribute to neurodegeneration, such as TNFR1, MMP9, ICAM-1, and TRAF3 showed an increase in expression from 48 to 168 h after TMT administration in transcriptomic data (Fig. 9, Online Resource 1, Online Resource 2). One of these genes is Tnfrsf1a, which codes for TNFR1, the primary receptor for TNF. Neuronal TNF receptors perform fundamentally different roles in CNS pathology in vivo, with neuronal TNFR1 and IKKβ promoting microglial inflammation and neurotoxicity in demyelination (Orti-Casan et al. 2019; Papazian et al. 2021). Another upregulated gene is MMP9, which has dual effects. It promotes neurogenesis, angiogenesis, myelogenesis, and axonal growth, while also causing destructive effects such as apoptosis, disruption of the blood–brain barrier, and demyelination (Kaminari et al. 2018). Interestingly, MMP9 has been linked to Alzheimer's disease in the gene-disease association results of Open Targets and DISGENET plus (Online Resource 3). Furthermore, ICAM-1, whose transcript levels increased from 48 to 168 h after TMT administration, showed a corresponding rise in protein levels from 72 to 168 h after TMT administration (Online Resource 1, 2). ICAM-1 contributes to neuroinflammation by inducing the expression of pro-inflammatory cytokines and chemokines (Walker et al. 2017). This gene was also associated with Alzheimer's disease in the gene-disease association results of DISGENET plus (Online Resource 4). In contrast, TRAF3 encoding for TNF Receptor-Associated Factor 3 exhibited a decrease in transcript levels from 12 to 168 h after TMT administration. TRAF3 is involved in regulating the calcineurin-NFAT pathway and NF-κB pathway, thereby preventing excessive inflammation (Wang et al. 2012). Therefore, the downregulation of TRAF3 may contribute to neurodegeneration. These results indicate that TMT-induced gene expression affects inflammation and apoptosis in different ways, leading to neurodegeneration like Alzheimer’s disease.
Looking at the chemokine signaling pathway in transcriptomic data, various chemokines such as Cxcl10, Cxcl12, Cxcl14, and Cxcl16 exhibited upregulation from 24 to 168 h after TMT administration, indicating their involvement in the chemokine signaling pathway (Fig. 7, Online Resource 1). These chemokines are known to play a crucial role in neuroinflammation and have been found to be elevated in individuals with Alzheimer's disease (Li et al. 2023; Zuena et al. 2019). This correlation is further supported by the gene-disease association observed between Cxcl19 and Alzheimer's disease in the results from DISGENET plus (Online Resource 3). These results indicate the importance of neuroinflammation and chemokines in TMT-induced neurotoxicity.
In the apoptosis pathway, the Caspase-3 transcript shows an increase from 48 to 168 h post TMT administration (Fig. 8, Online Resource 1). Caspase-3 plays a crucial role in the apoptosis pathway that leads to neural cell death and is also involved in the TNF signaling pathway (Fig. 8, Fig. 9). Studies have shown its enrichment in the brains of Alzheimer’s disease patients, particularly in the hippocampus during the early stages of the disease (Louneva et al. 2008; Plociennik et al. 2015). Similarly, the PARP1 transcript is also upregulated within the same time frame. PARP1 acts as a primary sensor protein for DNA damage, and its upregulation after DNA damage results in mitochondrial dysfunction, increased oxidative stress, and NAD + depletion, which have been observed in various neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, Huntington's disease, and ALS (Thapa et al. 2021). The CTSD gene, responsible for encoding lysosomal cathepsin D, is also upregulated not only in transcriptomic data (from 24 to 168 h after TMT administration) but also in proteomic data (168 h after TMT administration) (Fig. 7, Online Resource 1, 2). Studies have reported its upregulation in the neocortex of Alzheimer's disease, leading to neurofibrillary degeneration (Chai et al. 2018). This finding is further supported by the GDA results linking Alzheimer’s disease with CTSD (Online Resource 3). MCL1, which plays a role in the apoptotic/antiapoptotic balance of neurons (Mezache et al. 2015), is also upregulated from 48 to 168 h after TMT administration (Fig. 8, Online Resource 1) and has been associated with Alzheimer’s disease in GDA results (Online Resource 4). These results highlight the role of various genes in the apoptosis pathway in the neural cell death caused by TMT administration and their associations with a couple of neurodegenerative diseases.
Beyond the chemokine signaling, TNF, and apoptosis pathways, several other mechanisms contribute to neuro-axonal damage, including synapse loss, autophagy, dysregulated calcium homeostasis, and impaired axonal guidance. The observed increase in plasma NfL levels suggests progressive neuronal injury, which can be linked to these molecular disruptions.
One of these genes is SNAP25, which encodes synaptosome-associated protein 25. This protein plays a role in regulating synaptic vesicle fusion and neurotransmitter release (Hoerder-Suabedissen et al. 2018). The transcriptomic data indicates that SNAP25 is downregulated from 48 to 168 h after TMT administration (Online Resource 4). This downregulation can potentially contribute to the loss of synapses, ultimately leading to neuro-axonal damage as indicated by NfL levels. Another gene, Lamp1, encodes for lysosomal membrane protein 1 and is involved in the autophagic pathway (Lattanzi et al. 2013). Both transcriptomic and proteomic data show an upregulation of Lamp1 at 168 h after TMT administration (Online Resource 4). Additionally, the upregulated CTSD gene in the chemokine signaling pathway (Online Resource 1) is also implicated in calcium signaling (Lattanzi et al. 2013). In fact, calcium homeostasis is another affected pathway, closely linked to axon integrity. Calcium signaling (rno04020) ranked among the top 15 disrupted pathways, with over 18% of its genes identified as DEGs. Notably, five key calcium-related genes (ATP2B2, Adrb2, Ryr1, Ppp3cc, Prkca) were downregulated from 48 to 168 h post-TMT administration (Online Resource 1). Disruptions in calcium homeostasis impair neuronal function, synaptic transmission, and cytoskeletal integrity, ultimately contributing to axonal degeneration and NfL elevation (Nagendran and Taylor 2019).
These findings suggest that synaptic dysfunction, autophagy dysregulation, and calcium disturbances are major pathways contributing to neuro-axonal damage, reflected by increased plasma NfL levels.
Another critical pathway in TMT-induced neurotoxicity is axon guidance (rno04360), essential for maintaining neuronal connectivity and regeneration. Within 48 h post-TMT exposure, over 25% of its genes were differentially expressed, with eight key genes (Ntng2, Ntn3, Ppp3cc, Epha7, Ephb1, Pixna2, Sema6c, Wnt4, Camk2g) showing sustained downregulation from 48 to 168 h (Online Resource 1). Axon guidance disruption impairs neuronal repair and increases susceptibility to degeneration, likely contributing to elevated NfL levels as a marker of neuro-axonal damage.
Additionally, the neuroactive ligand-receptor interaction pathway (rno04080) is significantly affected by TMT exposure. At 48 h post-TMT administration, 12% of its genes were differentially expressed, with six genes (Chrm3, Kiss1r, Gabrg1, Pate10, Grin3b, Thrb) downregulated and seven genes (C3, C3ar1, Vgf, F2rl3, Calcb, Tspo) upregulated between 48 and 168 h (Online Resource 1). Among these, Gabrg1, which encodes the GABA-A receptor subunit gamma-1, plays a key role in GABAergic neurotransmission. Its downregulation could lead to altered inhibitory signaling, potentially contributing to excitotoxicity and neuronal damage, which in turn promotes axonal injury and increased NfL release (Cheng et al. 2017). Similarly, the downregulation of Grin3b, which encodes NMDA receptor subunit 3B, may impair glutamatergic signaling and synaptic plasticity, further weakening neuronal resilience to toxic insults (Kirtay et al. 2021).
Notably, Prkca and Gabrg1 are also part of the GABAergic synapse pathway (rno04727), reinforcing the role of inhibitory neurotransmission in TMT-induced neurotoxicity.
Collectively, these findings suggest that disruptions in GABAergic and glutamatergic signaling, combined with calcium homeostasis imbalance, autophagy dysregulation, and impaired axonal guidance, contribute to neuro-axonal damage following TMT exposure. The resulting synaptic dysfunction, neuronal stress, and impaired repair mechanisms likely drive the elevation of plasma NfL levels, reinforcing its role as a biomarker of axonal injury.
At 72 h after TMT administration, two genes in neurodegenerative disease pathways showed identical regulation patterns in both transcriptome and proteomic data (Fig. 6, Online Resource 4, Online Resource 7). Psmb3, which is elevated in both transcriptome and proteomic data, is an essential component of the ubiquitin–proteasome system (UPS), which aids in protein quality control and degradation pathways that are frequently disturbed in many neurodegenerative illnesses. Its overexpression may increase proteasome activity, resulting in faster destruction of damaged or misfolded proteins. Nonetheless, excessive degradation or dysregulated proteasome function caused by increased Psmb3 levels may also result in a cellular proteostasis imbalance, thereby aggravating cellular dysfunction or contributing to disease progression (Ding and Zhu 2018). Tubb4a, on the other hand, is downregulated across both transcriptome and proteomic datasets. Tubb4a is an important component of microtubules, which are responsible for cellular structure, intracellular transport, and division. Tubb4a mutations have been associated with hypomyelination and other neurological diseases, showing that it plays an important role in neuronal development (Duncan et al. 2017). It can also affect microtubule dynamics during stressful situations, making neurons more vulnerable (Liang et al. 2024).
In addition to the genes previously mentioned, over 1700 proteins exhibited significant changes exclusively in the proteomic data with no change in transcriptomic data. Among them, 11 proteins showed significant alterations between 72 to 168 h following TMT administration and displayed associations with Alzheimer's disease. These findings could be attributed to post-translational modifications impacting protein levels without affecting transcription. Among these 11 genes, 9 (APP, APLP1, APLP2, Hsd17b12, PLD3, PCOLCE, FABP5, ATP6AP2) were upregulated and 3 (RELN, PCP4, Ppp1r14c) were downregulated from 72 to 168 h post TMT administration. Upregulation of APP (Amyloid Precursor Protein) was reported to cause increased production of amyloid beta (Aβ) peptides, especially Aβ42, which are prone to aggregation and form amyloid plaques characteristic of Alzheimer’s disease. It has been associated with increased production of reactive oxygen species (ROS), which can cause oxidative stress and contribute to neurodegeneration. (Nithianandam et al. 2023; Zhou et al. 2011). APLP1 (Amyloid Precursor-Like Protein 1) and APLP2 (Amyloid Precursor-Like Protein 2) have been reported to be related to APP (Arvidsson et al. 2008). PLD3 upregulation has also been associated with lysosomal dysfunction and β-amyloid plaque formation (Nackenoff et al. 2021). ATP6AP2 encodes an accessory protein of the vacuolar H⁺-ATPase (V-ATPase) complex, which is crucial for lysosomal acidification and protein degradation. These results show that TMT-induced neurotoxicity can affect the proteins related to β-amyloid plaques formation in a post-translational manner, which then leads to neurodegeneration in a manner similar to Alzheimer’s disease. Furthermore, RELN, which is downregulated between 72 and 168 h after TMT administration, encodes a glycoprotein for the extracellular matrix that is essential for appropriate cortical layering in the brain and cerebellum during neurodevelopment. It contributes to the pathophysiology of Alzheimer's dementia (AD) by being a component of the apolipoprotein E (apoE) biochemical pathway (Seripa et al. 2008).
The examination of the relationship between TMT-induced gene expression and NfL levels, employing WGCNA on DEGs derived from transcriptomic data, revealed 1,377 genes in the black module that demonstrated a strong correlation with NfL, dose, and time, as well as 349 genes in the green-yellow module that correlated significantly with NfL levels. This represents about 29% of the differentially expressed genes in TMT-treated rats (Fig. 11a, Fig. 11b). The positive correlation values between module membership and gene significance for NfL in the black (Fig. 11c) and green-yellow (Fig. 11d) modules highlight the potential of these genes in targeting NfL. In these modules, the driving genes were identified as 397 in the black module and 57 in the green-yellow module, with thresholds set at MM > 0.8 and GS > 0.5. This represents roughly 6% of the total differentially expressed genes (DEGs). Among the 454 genes analyzed, 450 were found to have associations with Alzheimer’s disease.
Furthermore, we identified Hck from the chemokine signaling pathway (Fig. 7), Mcl and Ctsd from the apoptosis pathways (Fig. 8), and Tnfrsfla from both apoptosis and TNF signaling pathways, all of which exhibited upregulation from 48 to 168 h post-TMT administration. The findings demonstrate a correlation between TMT-induced gene expression and neuro-axonal damage, as evidenced by NfL levels, and further substantiate the link between TMT-induced neurotoxicity and Alzheimer’s disease.
The results also point to several areas for further study in the fields of neurotoxicity and neurodegeneration. A list of possible targets for therapeutic approaches is suggested by the 450 driving genes in the black and green-yellow modules that exhibit GDAs with Alzheimer's disease. Specifically, the identified driving genes in the TNF signaling pathways (Tnfrsfla), chemokine signaling (Hck), and apoptosis (Ctsd, Mcl) provide therapeutic targets for neuroprotective measures. Additional functional studies are necessary to confirm the significance of identified molecular targets and pathways in human tissue samples or cell models, as well as to validate potential therapeutic targets. Furthermore, examining cell type-specific responses to TMT administration using techniques such as single-cell sequencing may provide comprehensive insights into the mechanisms of neurotoxicity.


Comments (0)