In 1868, Paul Langerhans, as a medical student, described an epidermal cell population with characteristic dendrites using a novel “gold colloid” stain. Langerhans referred to these cells as extra-cutaneous nerves [7]. Over a century later and with the advent of electron microscopy, Langerhans’ neural doctrine was challenged and it was shown that LC are a specific subset of dendritic cells that play a pivotal role in immunity [8]. The ultrastructural hallmark of epidermal LC is the presence of cytoplasmic pentalaminar membranous granules, termed Birbeck granules. The Birbeck granules have a tennis-racket shape and are 200–400 nm long and 33 nm wide, with a zipper-like appearance [9]. The granules originate from cellular membranous structures that are internalized during the process of endocytosis [8]. Epidermal LC express Langerin (CD207), a lectin needed for the production of Birbeck granules [10]. More recently, the immunoexpression of Langerin (CD 207) has mainly replaced transmission electron microscopy for identification of LC.
LCH is defined by World Health Organization (WHO) as clonal neoplastic proliferation of Langerhans-type cells [11]. LCH is a rare disease. Over 50% of the LCH cases are diagnosed in patients under 15 years of age. LCH is extremely rare in adults (1 case per million per annum) [12]. Clinically, LCH is classified according to the number of involved organs. Systemic involvement is classified as unisystemic or single system (involvement of a single organ) or multisystemic (two or more organs involved); within each organ the disease is categorized as unifocal or multifocal (one or more than one lesion, respectively) [12]. Bones are frequently affected, followed by the skin. Head and neck manifestations of LCH are frequent, with reported frequencies varying from 50–80%.13 In a large case series, 54% of patients had craniofacial osseous involvement, which included the calvaria, skull base, temporal, and maxillofacial bones [13]. Following calvaria and usually affecting 10% of LCH patients, the mandible is the second most common bone involved in LCH [13]. The posterior region is the most common location for mandibular lesions [12]. The gingival tissues are frequently affected in patients with gnathic LCH, with reported frequencies of up to 20%.13 LCH involving only soft tissues may also occur, particularly the gingiva and hard palate [12].
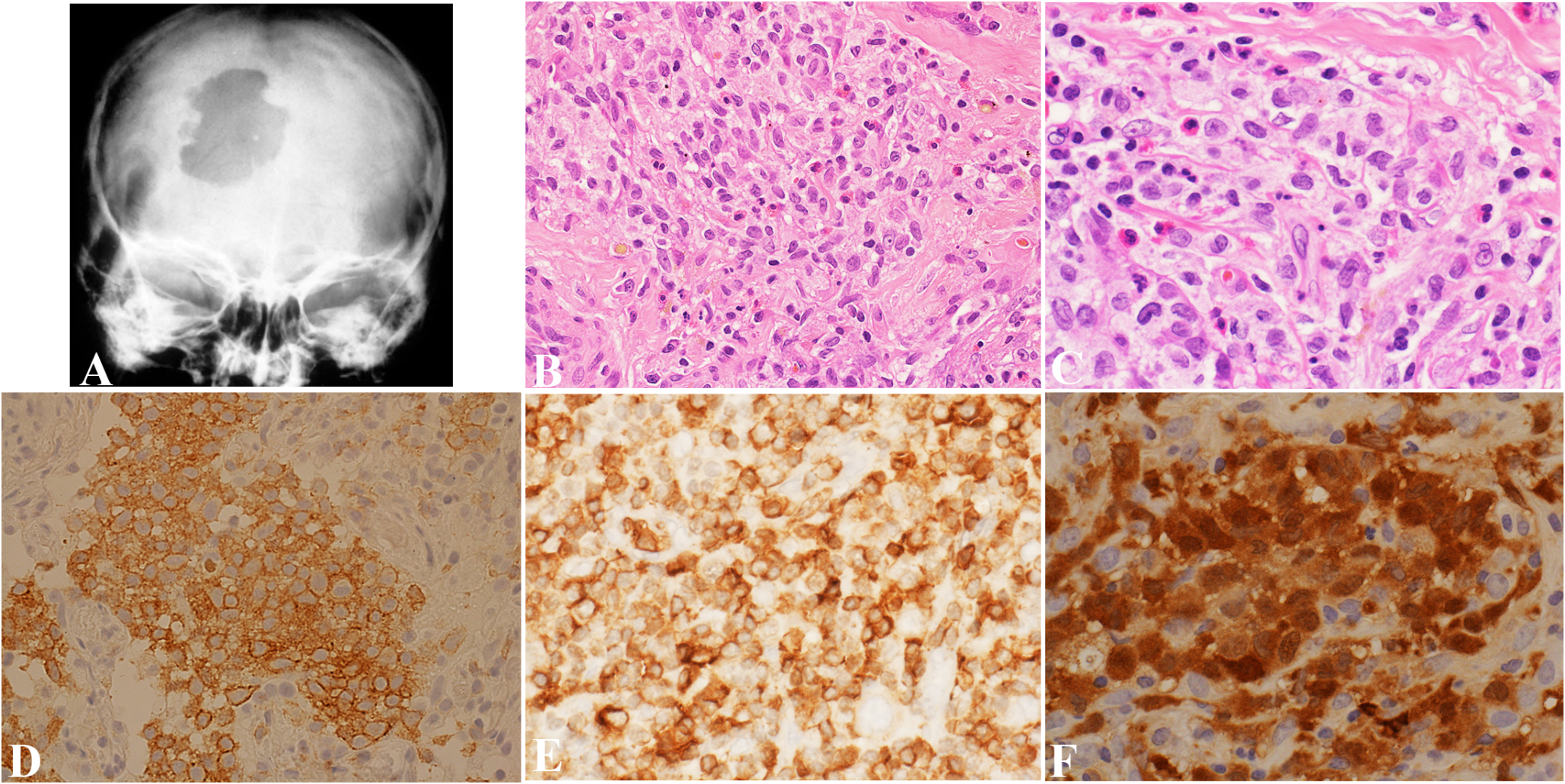
Radiologically, LCH may present as a unilocular or multilocular radiolucency (Fig. 1a) with bone loss and floating teeth [14]. Periapical presentations of LCH are common [14, 15]. In a study of 118 cases of non-endodontic periapical lesions, squamous cell carcinoma was the most predominant malignancy misdiagnosed as apical periodontitis on radiological examination (n = 7, 5.93%), followed by adenoid cystic carcinoma (n = 1, 0.85%), and LCH (n = 1, 0.85%) [16].
Subtle radiological signs of an aggressive lesion in an apparently benign appearing radiolucency should alert to the possibility of a locally aggressive or malignant lesion.
Cases with multiple punched out lytic lesions of the skull closely resemble multiple myeloma [17]. Skin lesions are characterized by erythematous eruptions or a discreet pinkish-brown papule [18].
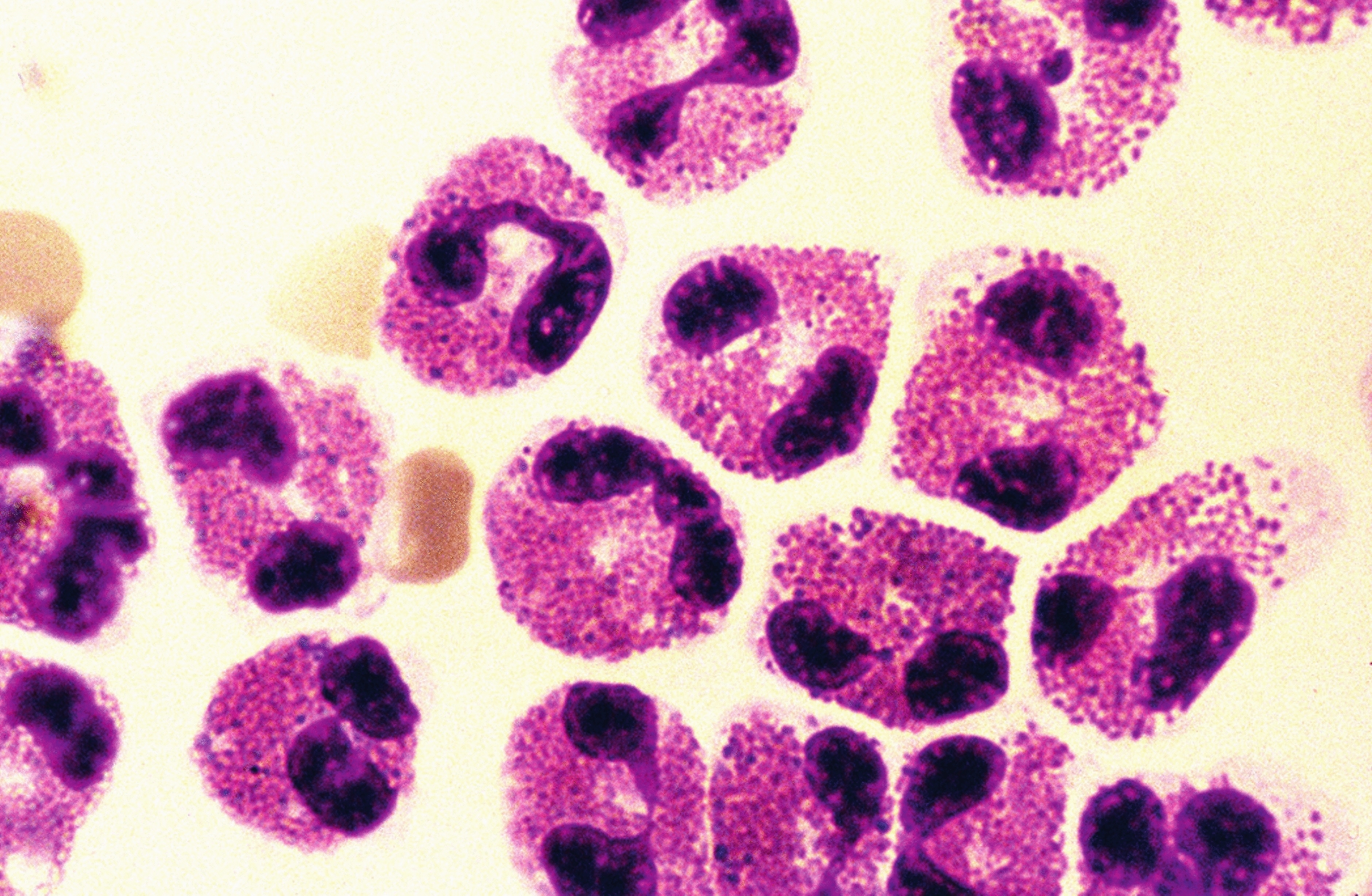
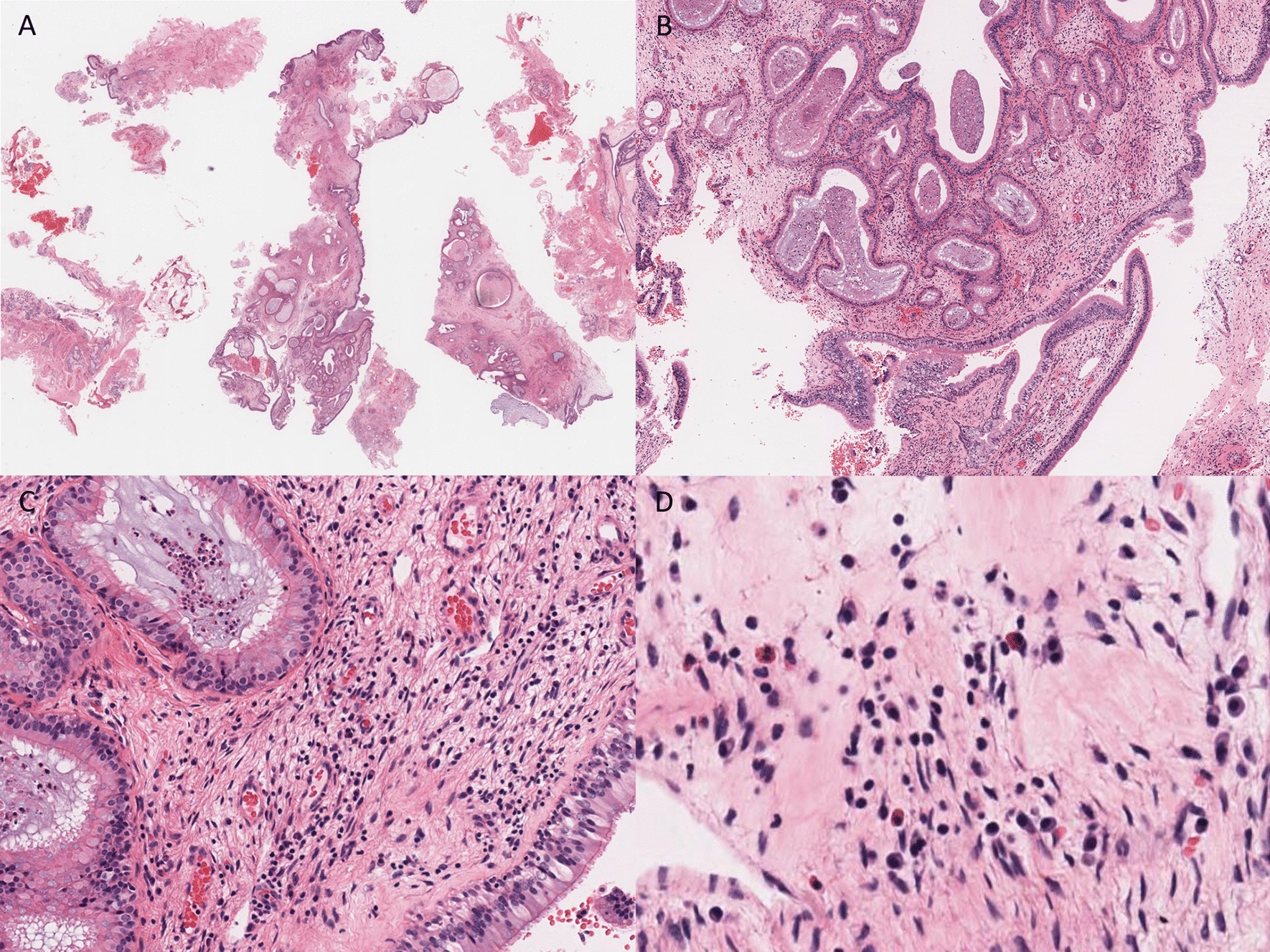
Histologically, a solid infiltrate of cells with grooved folded nuclei (coffee bean-shaped nuclei) is seen in LCH. The reactive inflammatory component consists of eosinophils. In addition, neutrophils, lymphocytes, macrophages, plasma cells, and multinucleated giant cells may also be seen [19]. Atypical mitoses and apoptotic bodies have also been described, with necrosis observed in cases with a high proliferation index [19]. In one study, a Ki-67 proliferative index of > 40% was correlated with the presence of multisystem disease [19].
LCH shares surface markers with epidermal LC (CD1a+/CD207+).4 Histomorphological and immunohistochemical similarities between LCH cells and epidermal LC have resulted in the persistent view that LCH is a disorder of transformed or activated epidermal LC. More recently, dendritic-cell subsets have been discovered with the potential to express Langerin (CD 207) and form Birbeck granules, that survey tissues with increased recruitment from blood to tissue during inflammation [20]. Badalian-Very and colleagues have detected BRAF p.V600E in 57% of LCH lesions [21]. In addition to BRAF p.V600E, other activating mutations in BRAF, including in-frame deletions, fusions, and duplications, have been identified in LCH lesions [4]. Thus, LCH cells are more likely to arise from dysregulated differentiation of bone marrow–derived precursor cells with activating MAPK somatic mutations rather than from transformed or activated epidermal LC [4]. The latter is known as the “misguided-myeloid-differentiation” hypothesis [4]. Similarly, we hypothesize that, the chronic inflammatory microenvironment in a periapical cyst encourages active recruitment of genetically altered Langerhans-type cells from blood to the connective tissue capsule of the cyst, in a patient with established LCH.
A deeper insight into the evolution and differentiation of dendritic cells, may help better understand the differential diagnosis and immunoexpression of LCH.
Monocytes characteristically express CD163, S100, and OCT2 [22]. As they migrate into tissues and assume a macrophage phenotype, they exhibit enhanced expression of CD163 and Factor XIIIA with low expression of S100 and OCT2 [22]. Alternatively, if monocytes differentiate toward a dendritic cell phenotype, they show decreased expression of CD163 and OCT2. Indeterminate dendritic cells that are presumed to be LC precursors, express S100 protein, CD1a, and CD4, but are negative for Langerin (CD 207); interdigitating dendritic cells express S100 protein and CD4, but are negative for CD1a and Langerin (CD 207) [22]. Differentiation toward a mature LC phenotype is demonstrated by expression of CD1a and Langerin (CD 207) [22]. Therefore, the histological differential diagnosis of LCH includes granulomatous inflammatory disease (e.g. tuberculosis or fungal infections) and neoplasms in the same line of differentiation as LCH, namely, Langerhans cell sarcoma (LCS), interdigitating dendritic cell sarcoma, and indeterminate dendritic cell tumor (IDCT).
Coffee bean-shaped nuclei are not specific to LCH and may be seen in the setting of inflammatory conditions such as granulomatous inflammation. Careful attention to the arrangement of cellular components (granulomas) and IHC helps exclude granulomatous inflammatory disease. Distinction from LCS and IDCT may be challenging as both lesions show CD1a and S100 expression [22]. In addition BRAF p.V600E has been reported in LCS and IDCT [23].
In contrast to LCH, LCS is an aggressive malignancy, in which the Langerhans-type cells possess high-grade large and markedly pleomorphic round to oval vesicular nuclei with clumped chromatin and prominent nucleoli. Focally, some cells may exhibit the complex grooves of LCH cells, a clue to diagnosis, however, in general the diagnosis of LCS is challenging and often requires an extensive panel of immunohistochemical markers to exclude tumors with high-grade nuclei, including carcinoma, melanoma, lymphoma, and sarcoma [3, 11, 24]. The immunophenotype of LCS is identical to that of LCH, positive for CD1a and Langerin (CD 207) [3, 11, 24]. The percentage cut-off in the expression of CD1a and Langerin (CD 207) to establish a diagnosis of LCS is not defined by the WHO. One study has suggested strong CD1a and Langerin (CD 207) expression in more than 30% of LCS cells [22]. LCS shows variable eosinophil infiltrates and high mitotic activity, with more than 50 mitoses per 10 high-power fields [11].
IDCT is defined by the WHO as neoplastic proliferation of spindled to ovoid normal indeterminate cells, the presumed precursor cells of LC [25]. The IDCT cells resemble LCH cells with irregular nuclear grooves and ample eosinophilic cytoplasm and express dendritic cell markers, CD1a and S100. However, they lack the Birbeck granules of LCH on ultrastructural examination and do not express Langerin (CD 207) [3, 25]. The mitotic rate is variable from case to case and there is usually a mixed inflammatory cell infiltrate consisting of eosinophils, small reactive lymphocytes, macrophages, and multinucleated giant cells. IDCT are negative for the histiocytic marker, CD163 and follicular dendritic cell markers CD21, CD23 and CD35 [3, 25].
Interdigitating dendritic cell sarcoma is characterized by the proliferation of spindled to ovoid cells showing interdigitating dendritic cell phenotype [3, 26]. Unlike LCH, solitary lymph node involvement is the most common clinical presentation, however, extra-nodal involvement (skin and soft tissue) have been reported [26]. The tumor cells display abundant eosinophilic cytoplasm, indistinct cellular borders, and spindled to ovoid indented nuclei with small conspicuous nucleoli. Mitotic activity is low, with less than 5 mitoses per 2 mm [2] and Ki-67 proliferative index of 10–20%.26 Multinucleated tumor cells are occasionally seen. Different from LCH, interdigitating dendritic cell sarcoma grows in sheets, fascicles and whorls. Small lymphocytes are interspersed throughout the tumor. The tumor cells consistently express S100 protein and vimentin and are negative for CD1a, Langerin (CD 207), CD21, CD23 and CD35 [11, 22].
BRAF IHC, using the monoclonal antibody VE1, has been validated to recognize a mutant BRAF p.V600E protein with a negative predictive value of 92%.6BRAF IHC is efficient for detecting the V600E genetic variant, but negative cases should be further evaluated by DNA-based techniques for other BRAF variants, which have also been shown to drive clinical benefit from BRAF inhibitors [6]. DNA-based real-time PCR based assays are widely available in many laboratories across the world, including pathology laboratories in developing countries. One such assay is the automated Idylla™ assay with a positive- and negative predictive value of 100% for BRAF p.V600E [6, 27]. While we failed to demonstrate a somatic BRAF p.V600E variant by IHC and PCR-based assay, the possibility of other activating mutations in BRAF, especially in-frame deletions cannot be entirely excluded. In fact, in-frame BRAF deletions are usually seen in patients with multisystem LCH compared to single system LCH, and especially in those with liver involvement with associated poor outcome [28]. BRAF deletions are resistant to “first-generation” BRAF inhibitors and are sensitive to mitogen-activated protein kinase kinase (MEK) inhibitors [28]. Thus, determination of BRAF deletions in LCH patients lacking BRAF p.V600E may have prognostic and therapeutic implications. A study of 31 adult patients with LCH found that unifocal oral involvement is significantly associated with multisystem disease [29]. A multidisciplinary approach to patient management should be adopted with thorough clinical and radiological examination, hematological tests (full blood count, platelet count, coagulation tests and liver function tests) and urine biochemistry [30]. Focal jaw lesions are best managed by curettage [29]. Intralesional corticosteroid injections, often help with pain management and encourage healing. The affected mucosal regions require periodontal treatment, ranging from prophylaxis to root planning, depending on the patient’s oral hygiene status. Tooth extractions should be restricted to areas with significant bone loss with lack of periodontal support [29]. The prognosis is good in most cases and the estimated three-year overall survival for adult patients is 94.4%.28 Liver or spleen involvement and age older than 50 years indicate poor prognosis [28].






Comments (0)