Trials included
This PopPK analysis included data from three preclinical studies in cynomolgus monkeys and two phase I/IIa clinical trials in SARS-CoV-2 infected and uninfected individuals receiving DZIF-10c treatment. An overview of these studies is provided in Supplementary Table S1.
In the first preclinical study, 6 anesthetized cynomolgus monkeys received single IV bolus (injection over 2–3 min, n = 3) or intratracheal (IT; instillation via microsprayer administered into the lumen of the trachea, n = 3) doses of DZIF-10c (50 and 3.6 mg/kg, respectively) (Boehringer Ingelheim, data on file). Serum and bronchoalveolar lavage (BAL) samples were collected regularly over 28 days (see Supplementary Table S1 for details). In the second study, 4 cynomolgus monkeys received repeated INH doses of DZIF-10c (through a nebulizer, ∼ 1000 mg per dose) or vehicle over 7 days (Boehringer Ingelheim, data on file). Plasma samples were collected over the duration of treatment. In the third study, 4 cynomolgus monkeys received two prophylactic INH doses of DZIF-10c (via nebulizer, 500 mg per dose) prior to infection with SARS-CoV-2 [16]. Plasma and BAL samples were collected at least daily. The protocol for the first monkey study was approved by the Animal Care and Use Committee (in compliance with the Animal Welfare Act Regulations [9 CFR Part 3]) of LabCorp, Madison, Wisconsin, USA. Protocols for the other two monkey studies were approved by the Animal Welfare Body of Cynbiose (Marcy l’Étoile, France) and the Ethics Committee of VetAgro Sup (Marcy l’Étoile, France). The animals received humane care according to the Association for Assessment and Accreditation of Laboratory Animal Care and the French regulation (dated February 7, 2013) implementing European Directive 2010/63/UE.
In the first phase I/IIa clinical trial, 57 individuals with and without a SARS-CoV-2 infection received a single IV infusion of DZIF-10c (registered at www.clinicaltrials.gov as NCT04631666 [17]). In the phase I open-label dose escalation part of this trial, 15 healthy subjects received single rising IV doses of DZIF-10c (2.5–80 mg/kg, infusion over ∼ 60 min) and 3 participants positive for SARS-CoV-2 received a 40 mg/kg dose of DZIF-10c within 7 days of onset of symptoms (Supplementary Table S2). In the subsequent phase IIa double-blind expansion part of the trial, 39 individuals who tested positive for SARS-CoV-2 were randomized to receive either a single dose of DZIF-10c (40 mg/kg, n = 26) or placebo (n = 13).
In the second clinical trial, 45 individuals with and without symptomatic SARS-CoV-2 infection received a single INH dose of DZIF-10c (50–250 mg, via a nebulizer) over 15–20 min (NCT04631705) [18]. This trial had a similar design to the first trial with a phase I, open-label dose escalation part in 9 healthy subjects and 9 participants positive for SARS-CoV-2, followed by a double-blind, randomized, placebo-controlled expansion part in 27 participants with symptomatic SARS-CoV-2 infection. The phase IIa part of the trial included a further group of participants randomized to receive either combined INH (250 mg) + IV (40 mg/kg) DZIF-10c (n = 9), INH (250 mg) DZIF-10c + IV placebo (n = 8), or INH + IV placebo (n = 10). PK data from the 17 individuals treated with DZIF-10c in this part of the study was not available at the time of analysis. Only data from 18 individuals in the phase I dose escalation phase of the trial were available for inclusion in the PK dataset analysis.
For both trials, the objectives were to evaluate the safety, PK profile, immunogenicity, and antiviral activity of DZIF-10c. Across both trials, SARS-CoV-2-infected individuals received DZIF-10c early during their infection; an average of four days (range 1–8 days) after their first positive SARS-CoV-2 test. Both trials were conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Written informed consent was obtained from all subjects before study entry.
PK sampling and bioanalytical assays
Blood sampling schema for measurement of DZIF-10c serum/plasma concentrations varied in the preclinical and clinical studies (see Supplementary Table S1). BAL samples were collected from the lower respiratory tract in two preclinical studies (the first and third). BAL is an established method for sampling ELF from the lung but requires correction for varying degrees of dilution of the ELF sample due to saline used during the BAL procedure [19]. The urea correction method described by Rennard and colleagues [20] is commonly used to account for the ELF dilution and to estimate drug concentrations in the BAL fluid and was utilised for conversions. In the first preclinical study, concentrations of urea in serum and the BAL fluid supernatant were quantified using commercial colorimetric and enzymatic assay kits. Using the serum-to-BAL fluid urea ratio method [20], ELF concentrations in BAL samples were determined. In the third preclinical study, although BAL samples were collected, they could not be converted to ELF as urea levels were not measured.
In the first preclinical study, serum and BAL fluid concentrations of DZIF-10c were determined in-house using a quantitative ligand binding ELISA assay to detect total drug IgG (anti-human IgG capture and anti-human IgG HRP detection), following assay calibration in serum and BAL fluid, thereby ensuring adequate assay performance (recovery, accuracy and precision). The lower limit of quantification (LLOQ) in serum and BAL fluid was 20 ng/mL. In the second and third preclinical studies, DZIF-10c concentrations in plasma samples were determined using non-GLP ligand binding assays targeting the Fc part of human IgG1 at two complementary epitopes. The lower limit of quantification for these studies depended on the sample dilution and was defined as the limit of detection (LoD) multiplied by the minimal dilution used for the corresponding samples. In human studies, serum/plasma concentrations of DZIF-10c were determined using a validated total human IgG assay at QPS (Newark, DE, USA). The LLOQ was 300 ng/mL in humans. All subjects with at least one quantifiable serum concentration were included in the analysis.
Population PK modeling
The PopPK analysis was performed using nonlinear mixed-effects modeling techniques (NONMEM, version 7.5, ICON Development Solutions, Ellicott City, Maryland, USA) (see Supplementary Material for model code). Population and individual model parameters were estimated using the first-order conditional estimation with η–ε interaction (FOCEI) method or the stochastic approximation expectation maximization (SAEM) method followed by importance sampling algorithm (IMP) during model development. Key and final models were run using the full Bayesian estimation method with four chains of 10,000 burn-in and 20,000 sampling iterations.
Data processing, visualization, and simulations were performed using R version 3.6.3 (www.rproject.org). Goodness-of-fit (GOF) plots, visual predictive checks (VPCs), and dose-exposure simulations were conducted using R and relevant packages used in pharmacometrics workflow such as mrgsolve (www.mrgsolve.github.io), vpc, and pmplots.
Structural model development
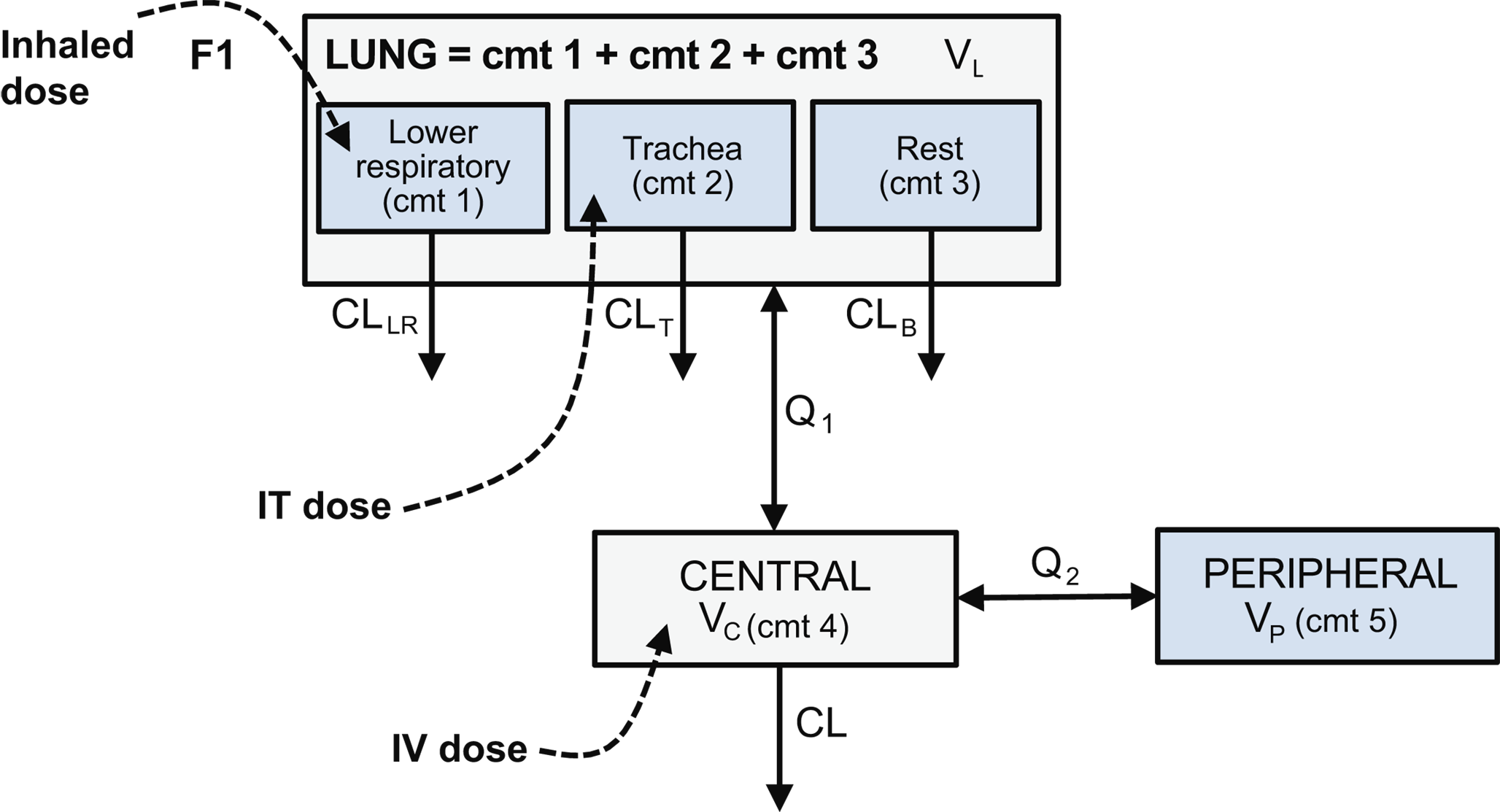
PopPK models were developed for DZIF-10c to describe exposure in the systemic circulation (plasma/serum) and lung (ELF), following single or multiple IV, IT, or INH, doses of DZIF-10c.
The population serum/plasma DZIF-10c concentration-time relationship displayed a typical bi-phasic PK profile for a mAb [21]. The systemic PK profile was well described by a two-compartment model with linear clearance. For INH administration, a first-order absorption from lung to systemic circulation was implemented.
Different models were tested to account for the physiological differences resulting from the different routes of administration. For this, the whole IT dose was assumed to be deposited in the lung compartment, while for the INH dose only a fraction of the dose was assumed to be deposited in the lung. DZIF-10c concentrations were observed in serum/plasma after IT and INH administration, reflecting drug distribution from the different sites of the respiratory system to the systemic circulation. DZIF-10c concentrations in the ELF compartment were also observed after the IV administration, reflecting the pulmonary distribution of drug via the systemic circulation.
In the first preclinical study, the inclusion of ELF concentrations after IV and IT administration in monkeys allowed characterization of the lung PK and informed a more mechanistic model with an ELF (lung) compartment. Therefore, a three-compartment model (lung-central-peripheral) was chosen as the basic model structure to describe plasma/serum and ELF PK data in the studies described.
As mentioned, the lack of measured urea concentrations in the third preclinical study meant the measured BAL concentrations could not be easily converted to ELF concentrations. To overcome this, different proportional models were implemented to correlate ELF and BAL concentrations. However, none of the models was able to simultaneously describe the data, indicating a more complex (e.g., non-proportional) relationship between ELF and BAL concentration. In the final analysis, BAL concentrations were not included in the analysis dataset.
Interindividual and residual variability
Nonlinear mixed effects models were developed to describe the PK data of DZIF-10c in the central and lung compartments. The approximate joint posterior distribution of estimated population parameters (i.e., covariance matrix of the estimates) follows a multivariate normal distribution. Most PK model parameters were modeled in the log domain to be consistent with this distributional assumption. Bioavailability parameters were modeled using a logit transformation to constrain the distribution between 0 and 1. For the parametric nonlinear mixed effects model, parametric distributions were assumed for the random effects.
Interindividual variability (IIV) was described using an exponential or inverse-logit error model for bioavailability. IIV was initially estimated for all PK parameters, with estimates showing high uncertainty fixed to 0.0225, as recommended for SAEM and BAYES estimation methods [22]. Residual (unexplained) variability (ε) for plasma/serum and ELF concentrations were described separately by a combined additive and proportional error model using the variance-covariance matrix of the residual random errors (see Supplementary Material for all model equations).
Covariate analyses
Due to the characteristics of this analysis (combining human and cynomolgus monkey data), the small size of the studies and the objective of the analysis, no demographic covariates were evaluated. Body weight dependent allometric scaling was used to explain differences in the volumes of distribution and clearance rates. Weight and species were also used to explain the difference in bioavailability between humans and cynomolgus monkeys after INH doses.
To allometrically scale from monkey to humans and explain the differences in exposure by the observed weight distribution, we standardized weight by the typical human value of 70 kg (weight/70) and scaled using fixed exponents of 0.85 for clearance rates and 1 for the different volumes of distribution without further adjustment of parameters [15].
Model selection and evaluation
Model selection was guided by numerical change in objective function values (OFV), physiological relevance of parameter estimates, precision of parameter estimates, minimization of Akaike information criterion (AIC), and overall GOF plots. GOF plots included diagnostic scatter plots, figures of normal prediction distribution error (NPDE) values versus time and population predicted values, and plots of observed concentrations versus population and individual predicted concentrations. Models run using the Bayesian estimation method were analyzed for convergence using trace plots and density plots of posterior parameter distributions. Final model parameter estimates were reported with a measure of estimation uncertainty including 95% credible intervals (CDIs) from the posterior parameter distributions of the Bayes runs.
The final model was qualified by inspection of longitudinal prediction-corrected visual predictive check (pc-VPC) [23], NPDE [24] plots and by Bayesian model fits [25]. In addition, the precision of the parameters was estimated via asymptotic standard errors of the estimates provided from the $COVARIANCE step in NONMEM. For the longitudinal pc-VPCs, parameter uncertainty was assumed negligible relative to inter-individual and residual variance [26, 27]. pc-VPCs were performed with Monte-Carlo simulations. Five hundred datasets, identical in structure to the original dataset, were simulated using the developed PopPK model. Plots of the observed concentrations in plasma/serum and ELF were then constructed and overlaid with the corresponding simulated median, 5th, and 95th percentiles at various time periods as appropriate. Finally, the precision of the final model parameters was investigated using the full Bayesian estimation method in NONMEM. Weakly informative priors were used, centered around parameter estimates from the final model developed using the SAEM-IMP analysis method [22]. Four chains of 10,000 burn-in and 20,000 sampling iterations in the accumulation phase were used. 95% confidence intervals (CIs) for the parameter estimates were generated by observing the 2.5th and 97.5th percentiles of the parameter estimates in the accumulation phase.
Model simulations
The developed model was used to perform different simulations (including IIV and residual variability) in humans after IV and INH administration. Simulations for putative clinical dosing regimens (INH single dose, IV single dose, and simultaneous INH and IV doses) were performed. Area under the concentration-time curve for 4 weeks after the dose (AUC4w), maximum concentration (Cmax), and trough concentrations 4 weeks after dose (Ctrough,4w) were calculated for the different simulations.
Simulated DZIF-10c exposure (ELF and serum/plasma concentrations) were compared with the 100% neutralizing concentrations (NC100) obtained from a live virus assay with relevant SARS-CoV-2 variants at the time of analysis [28]. For this comparison, the weakest (highest) NC100 at that time (the SARS-CoV-2 variant P.2 [first identified in Brazil] with an NC100 of 0.397 µg/mL) was used as the reference [28].
To inform the bodyweight covariate in the model, an in-silico population was derived from sampling (n = 1,000) using the NHANES data set (www.cdc.gov/nchs/nhanes/index.htm). Data sets for years 2017-18 were filtered to only include subjects aged ≥ 18 years and ≥ 40 kg in body weight. From this realistic weight/age distribution, 3,000 human subjects were simulated (1,000 per administration/dose route).



Comments (0)