
Remember me
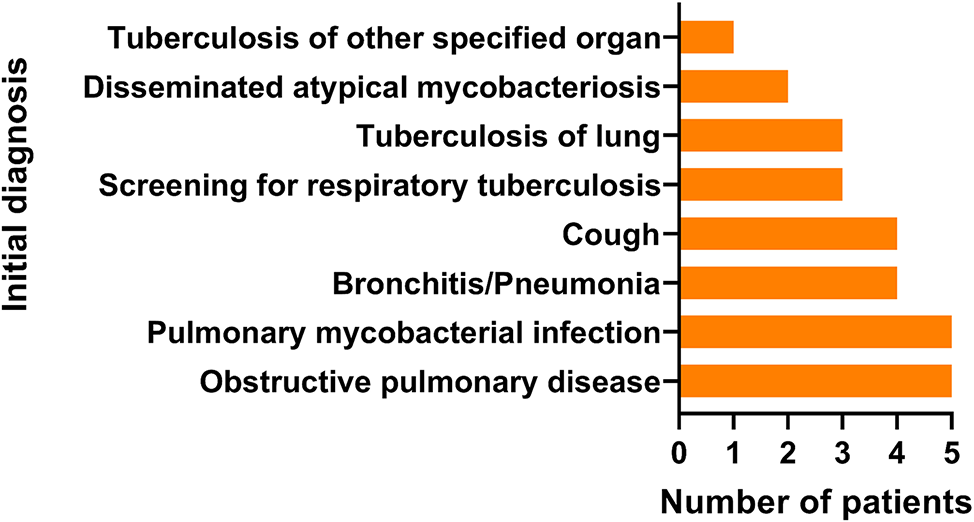
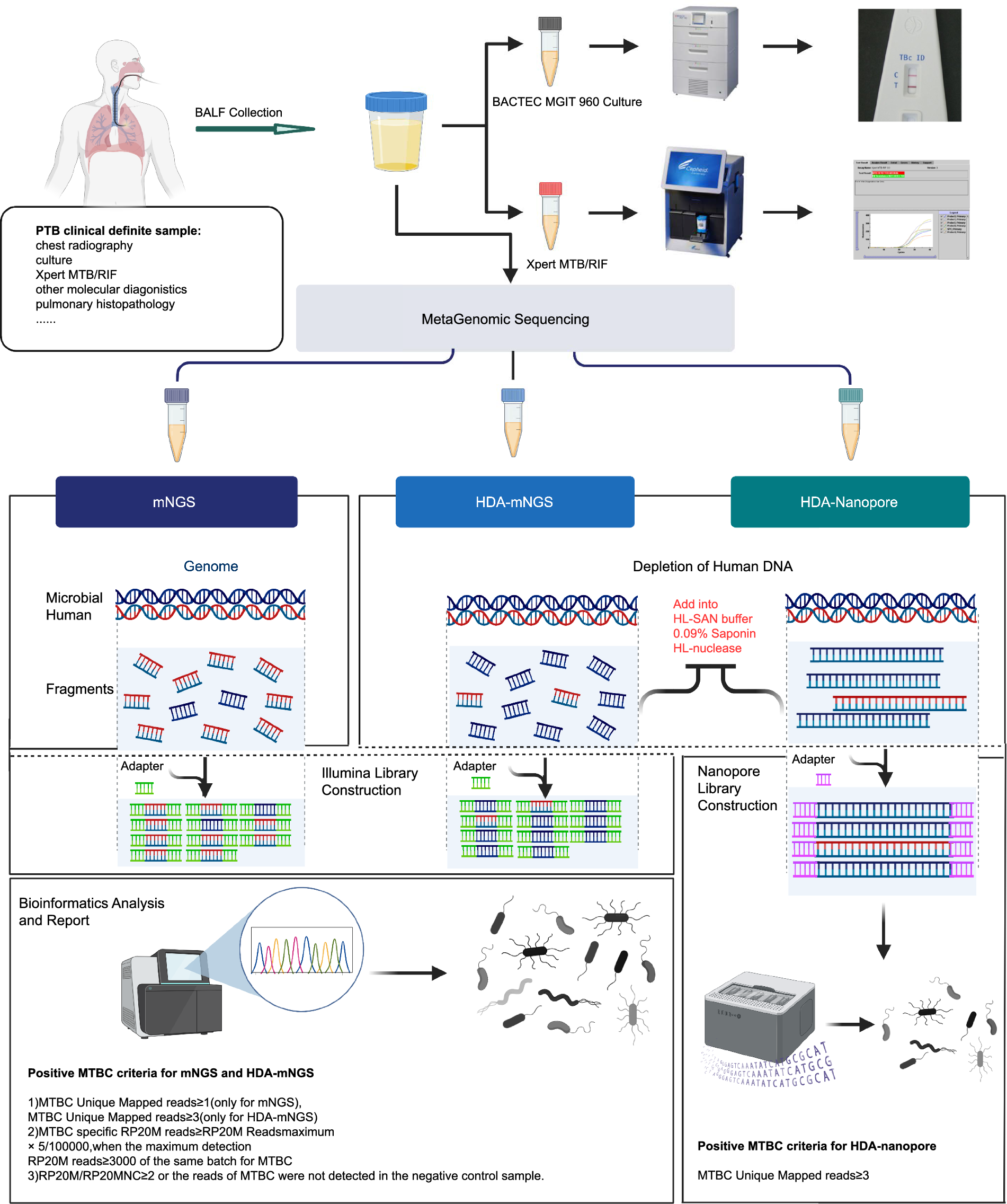





Between June 1, 2021, and May 1, 2022, a total of 105 patients with clinical or radiological suspicion of active pulmonary TB were screened from three hospitals for review and prospective enrolment in this study. Tuberculous tests, including culture, Xpert, mNGS, HDA-mNGS and HDA-Nanopore, were conducted on BALF samples from 105 patients (Fig. 1). Out of 105 samples, 7 patients who met our exclusion criteria were excluded for analysis. The average age of the remaining 98 patients (57.1% male) was 46 (Table 1). Of these patients, 28.6% (28 patients) had underlying pulmonary disease, such as chronic obstructive pulmonary disease (COPD), bronchiectasis, and lung cancer, while 24.5% (24 patients) had underlying extrapulmonary disease. Acid-fast bacillus (AFB) smear by microscopy was positive in 17.4% (17 patients) of the patients.
Fig. 1
Flowchart showing steps of MTB-detection methods used in this study. BALF samples collected from patients were each aliquoted into five tubes that were subjected to separate tests that included MGIT 960 culture (one sample), Xpert (one sample) and three mNGS tests (three samples) that included conventional mNGS (one sample), HDA-mNGS (one sample) and HDA-Nanopore (one sample). The conventional mNGS library was constructed without depletion of human DNA, whereas MGI and Nanopore libraries were constructed after human DNA was removed from the HDA-mNGS and HDA-Nanopore pipelines. Sequencing, bioinformatic analysis and reporting PTB diagnostic results were interpreted based on standard TB cut-offs. The mean sequence number outputs obtained using conventional mNGS, HDA-mNGS and HDA-Nanopore were 56.84M, 28.16M and 103.74K. Created in BioRender [16]
Table 1 Demographic and Clinical Characteristics of the 98 PatientsOf the 98 patients, 83.7% (82 patients) were finally diagnosed with active pulmonary TB infection, including 50 patients with definitive results of PTB pathogen detection and 32 patients with clinical diagnoses (Additional file 1: Fig. S1). The remaining 16.3% (16 patients) were diagnosed with non-TB disease, including one case of nontuberculous mycobacteria (NTM), three cases of common bacterial infection, two cases of viral infection, three cases of fungal infection, one case of acute exacerbation of COPD (AECOPD), one case of diffuse panbronchiolitis (DPB), one case of Mycoplasma pneumoniae infection, and two cases of malignant lung disease (Additional file 2: Table S1).
HDA-mNGS increases both microbial and TB reads in BALF samplesAfter quality control and exclusion of non-biologically significant signals, 97.2% of the 98 conventional mNGS samples were identified as sequences of human origin. Following the removal of host reads, only 0.24% of the sequencing data remained, attributed to microbial genomes based on the microorganism genome database (unpublished). As a contrast, following the host DNA depletion procedure, both HDA-mNGS and HDA-Nanopore exhibited marked improvements. The average percentage of host reads decreased to 39.7% and 27.4%, respectively, while the average percentage of microbial reads increased significantly to 18.8% and 61.1%, respectively. This underscores the enhanced efficacy of the host DNA depletion method (Fig. 2A, B).
Fig. 2
HDA-mNGS increases both microbial and TB reads in Xpert positive samples. A Boxplot of host reads rates from three methods suggest that HDA steps reduced human DNA percentage rates to thereby dramatically increase the Pathogen Reads rate, as confirmed using the Wilcoxon test. B Metagenomics sequencing methods that incorporate steps to substantially remove host DNA were associated with increased Pathogen Reads rates. C The Ct value of Xpert indicated a negative correlation with the number of normalised reads obtained by metagenomic sequencing. D According to Ct value cutoffs (< 22, 22–28, > 28), the detection performance of metage-nomic sequencing gradually decreased with decreasing MTB DNA content. Moreover, HDA-mNGS results were more robust as compared to results obtained using mNGS and HDA-Nanopore: Coverage of the M. tuberculosis genome obtained using metagenomic sequencing, based on Ct values for different patient groups (non-PTB versus PTB groups). The results indicated that HDA-mNGS provided excellent coverage depth that exceeded the coverage depths obtained using the other two methods
We next sought to determine whether the increase of microbial reads could proportionally yield an increase in TB reads. To this end, we utilize the Ct values from Xpert tests to determine the correlation between Ct values and the read numbers obtained through metagenomic methods. Among the 39 Xpert-positive samples, Ct values were obtained for 37. A negative correlation was identified between the Ct value and the normalized read number of MTBC using three metagenomic methods (Spearman correlation index: vs. mNGS R2 = 0.47, p < 0.001; vs. HDA-mNGS R2 = 0.54, p < 0.001; vs. HDA-Nanopore R2 = 0.58, p < 0.001) (Fig. 2C).
Following the cutoffs from a previous study [14], samples were categorized into three groups based on Ct values (Medium: Ct < 22, Low: Ct 22–28, Very low: Ct > 28). The MTBC detection performance exhibited a gradual decline with the transition of Ct values from the medium to the very low group (Fig. 2D). In the three groups HDA-mNGS outperformed mNGS in the MTBC reads (Wilcoxon test, p < 0.001), indicating superior pathogen detection performance. In Ct < 22 and Ct 22–28 groups, HDA-mNGS detected a high level of MTBC reads in all the 27 samples, whereas mNGS obtained reads in only 19 samples. In situation with poor sample quality (Ct > 28), HDA-mNGS still performed robustly, detecting reads in 9 out of 11 samples, compared to mNGS (4 out of 11) and HDA-Nanopore (6 out of 11). Furthermore, HDA methods (HDA-mNGS and HDA-Nanopore) significantly increased MTBC genome coverage in the medium and low groups, and with a nonsignificant increase in the very low group.
HDA-mNGS improves PTB diagnostic performance in BALF samplesWe evaluated the diagnostic performance of Culture, Xpert, mNGS, HDA-mNGS, and HDA-Nanopore for 98 BALF samples using sensitivity, specificity and accuracy based on the clinical final diagnosis (Fig. 3A, Additional file 1: Fig. S1).
Fig. 3
HDA-mNGS improves PTB diagnostic performance for BALF samples. A MTB detection rate obtained using five different methods. np, number of patients. B Heatmap of P value analyzed using McNemar test between HDA-mNGS with other methods (culture, Xpert, mNGS and HDA-Nanopore), * indicates the significance. (C) Comparative diagnostic performance of five different methods
Overall, as is expected, the application of HDA-mNGS has demonstrably enhanced the diagnostic efficacy of PTB in the present study. HDA-mNGS accurately identified 59 instances as positive for the MTBC, attaining a sensitivity of 72.0% (95%CI, 60.8–81.0%). According to McNemar test, the sensitivity of HDA-mNGS was significantly higher than the other four methods (Fig. 3B). Furthermore, HDA-mNGS exhibited the highest accuracy among the evaluated techniques, reaching 74.5% (95%CI, 65.9–83.1%). The performance of HDA-Nanopore ranked second in both sensitivity (58.5%) and accuracy (62.2%). Conventional mNGS and Xpert exhibited similar diagnostic performance, with sensitivities of 51.2% versus 49.4% and accuracy of 58.3% and 56.0% (Fig. 3C). Notably, as the currently golden standard for PTB pathogen detection, mycobacterial culture yielded positive results in only 25 samples. The culture method exhibited limitations, with 52 false-negative outcomes.
In the subset of definitively diagnosed PTB cases (n = 50) characterized by clear clinical manifestations, HDA-mNGS displayed a correct detection of 86.0% (43/50), surpassing the performance of conventional mNGS, which exhibited a rate of 54.0% (27/50). In the clinically diagnosed PTB cases, both mycobacterial culture and Xpert failed to detect MTBC. HDA-mNGS and conventional mNGS demonstrated comparable positive detection rate in detecting MTBC. However, they exhibited variations in the specific positive samples identified. In summary, the study highlights the significant improvement in diagnostic accuracy provided by HDA-mNGS for PTB.
Interestingly, we have investigated the co-infection among 98 specimens using metagenomic sequencing methods including conventional mNGS, HDA-mNGS, and HDA-Nanopore. Positive co-infection cases were defined as those detected by any of two methods. Out of the 82 PTB positive patients, 56 were found to have co-infections such as bacteria, fungi, and viruses, and HDA-mNGS demonstrated the best detection performance in co-infection analysis due to its highest rate (100.0%) of pathogen identification (Additional file 3: Table S2).
HDA-mNGS increases the coverage of MTB genome and drug resistance geneMultiple TB drug resistance-inducing mutations can be detected simultaneously by metagenomic sequencing but are highly dependent on sequence coverage. As reported previously, the detection rate of antimicrobial resistance genes (ARGs) using mNGS was extremely low due to limited coverage and depth of the MTB genome sequence [3]. In our study, 43 MTB positive cases using mNGS had a mean coverage rate of ≥ 1X across the MTB reference genome (H37Rv), reaching 95,267 bp (2.16%, ranging from 50 bp (0.001%) to 1,441,635 bp (32.7%) (Fig. 4A). In contrast, HDA-mNGS detected 61 MTB-positive cases with a mean coverage rate of > 1X across the MTB reference genome, reaching 1,528,168 bp (34.6%, ranging from 146 bp (0.003%) to 4,334,162 bp (98.2%)) (Fig. 4A). The sequences identified using conventional mNGS and HDA-mNGS methods varied across the different samples.
Fig. 4
Sequencing depth of MTB genome sequences obtained using mNGS and HDA-mNGS. A Coverage depth distribution of MTB genome. From inside to outside shows sequence depths of conventional mNGS of 1X and 3X and HDA-mNGS of 1X and 3X. The vertical text on the right indicates percent values of samples, whereby the circled numbers represent positions within the M. tuberculosis H37Rv sequence. B Heatmap of depth for each sample in 184 antimicrobial resistance locus
We explored the depth of 184 antimicrobial resistance locus published by WHO within the positive samples classified by mNGS and HAD-mNGS. The depth analysis showed that 15 of the 61 cases obtained from PTB cases had coverage depths of at least 3X, while 9 within these cases had had coverage depths of 10X or more (Fig. 4B).
In contrast, conventional mNGS provided only little sequence information compared to HDA-mNGS, making it impossible to determine the AMR gene coverage (Additional file 4: Table S3). Taken together, these results suggest that HDA-mNGS provided a greatly improved coverage of the MTB genome sequence and relatively higher AMR gene coverage in positive PTB cases.
Comments (0)