
Remember me
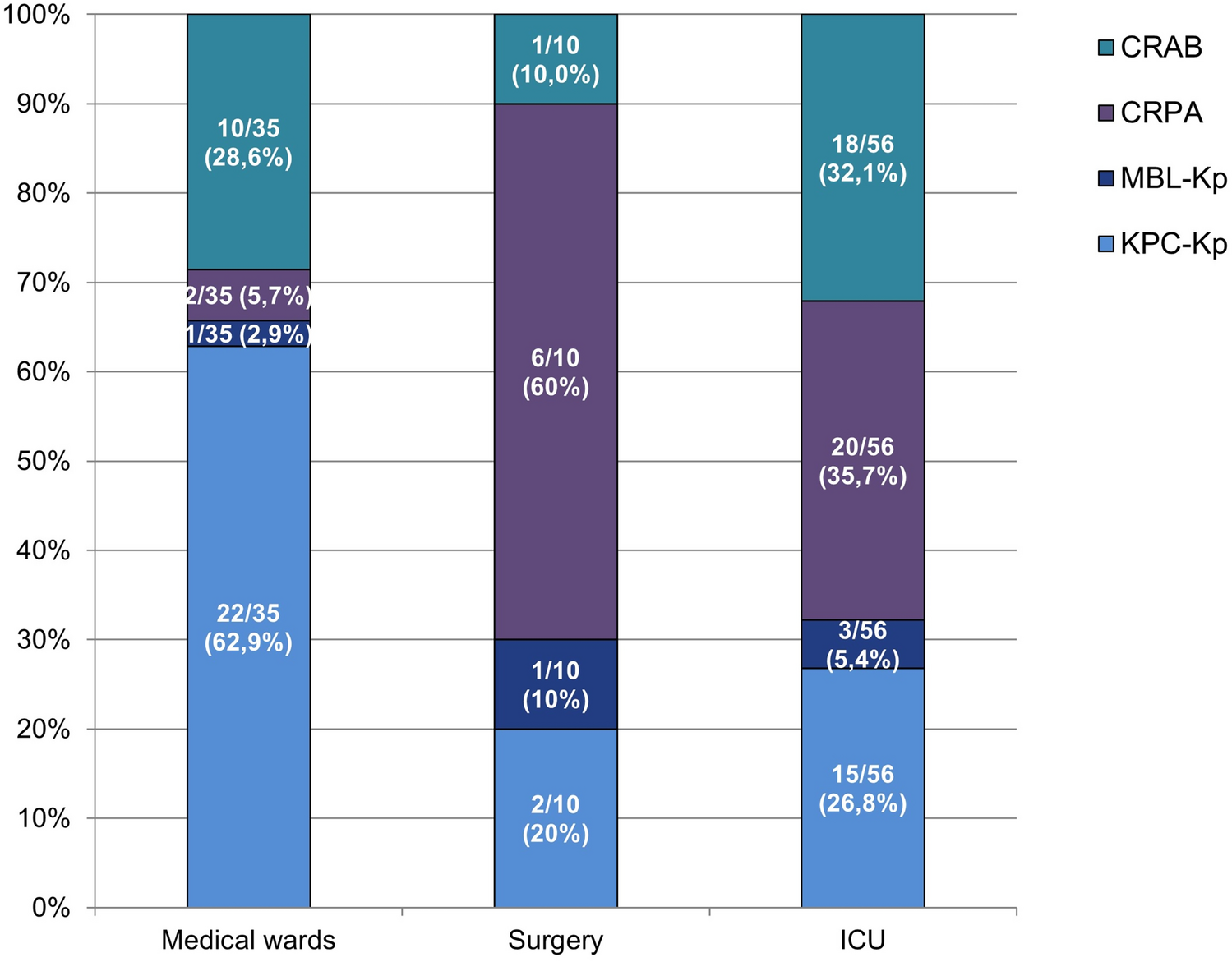
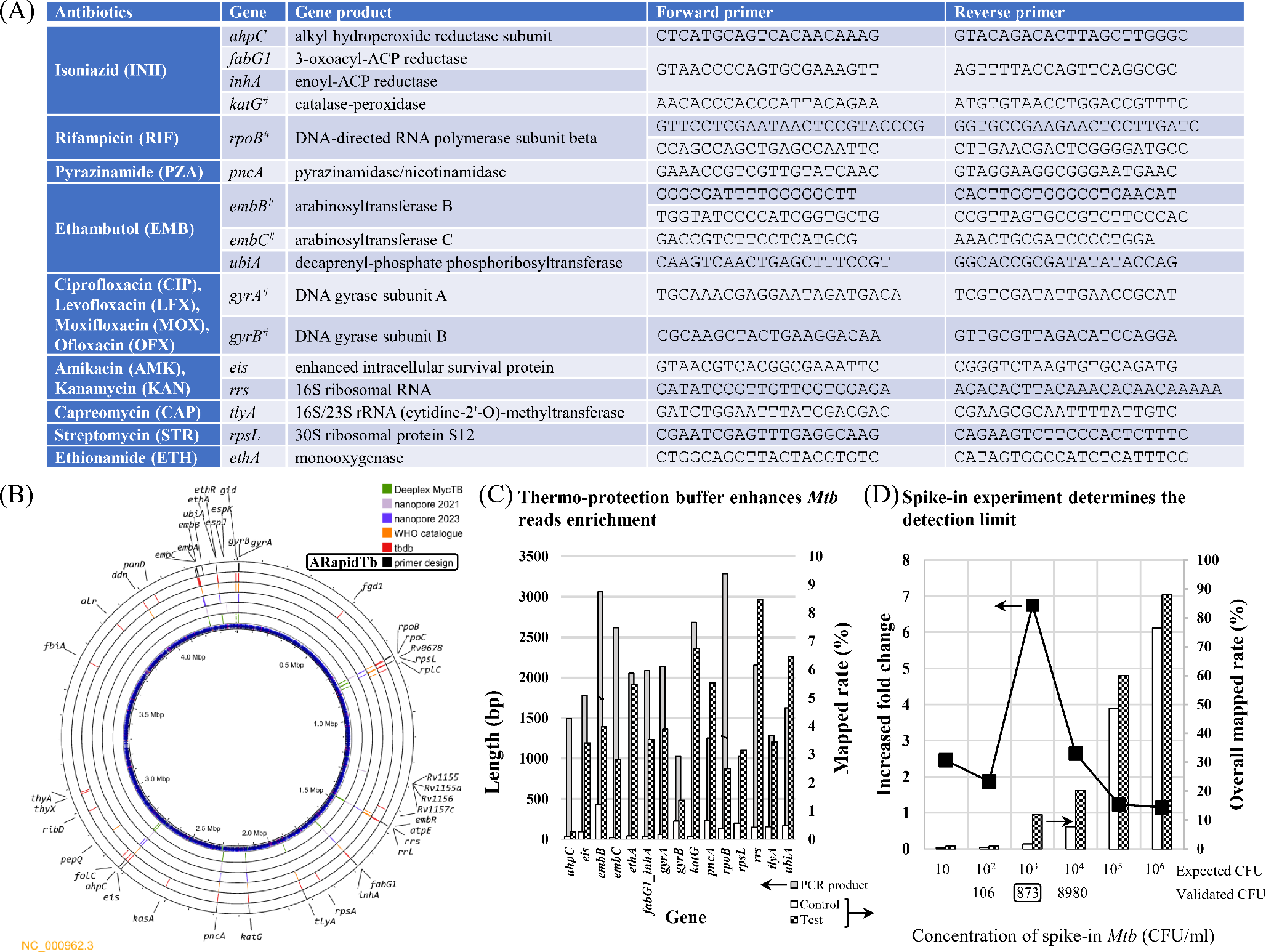
We extracted variants related to (a) amino acid substitutions, (b) SNPs in non-coding RNAs, and (c) SNPs in gene promoters from tbdb (the database of TBProfiler [22]) and from the WHO mutation catalogue [20,32] to compile the resistance-associated genes, as illustrated in Fig. 1B. We identified thirty-six resistance-associated genes containing 888 variant sites. While katG, pncA and ethA have more than 100 variant sites, alr, atpE, ethR, fbiA, fgd1, kasA, pepQ, ribD, rplC, and thyX have fewer than 5 sites. Variant-covering regions for these genes range from 1 bp to 2853 bp for thyX and embB, respectively. The resistance-associated mutations collected from both the tbdb and the WHO catalogue were saved as a Python dictionary named Tbresdb (https://github.com/jade-nhri/ARapidTb/blob/main/Tbresdb.txt). This database serves as a complication of resistance-associated mutations specifically designed for use in ARapidTb to facilitate resistance prediction. To compare the variant sites with the gene regions targeted by Deeplex-MycTB, we found some variants located in gyrB, rpoB, inhA, katG, ahpC, embB and ethA would be missed by Deeplex-MycTB (Supplementary Table S1). While Deeplex MycTB has been validated for use with nanopore sequencing on sputum samples [10], it covers only 13 kbp of genomic regions and does not fully exploit the advantage of long-read sequencing due to its short average amplicon length of 628 bp [12]. Recently, “nanopore 2021” [14] and “nanopore 2023” [15] introduced PCR assays that produce total product lengths of 33 kbp and 27 kbp, respectively, targeting nine and thirteen resistance genes (Supplementary Table S1). In addition to the ten common resistance-associated genes shared by Deeplex-MycTB, nanopore 2021, and nanopore 2023—gyrB, gyrA, rpoB, rrs, fabG1, inhA, katG, pncA, eis, and embB—this study further included rpsL, tlyA, ahpC, ethA, embC, and ubiA [33, 34] in the primer design to expand coverage for other second-line drugs, including capreomycin (CAP in Fig. 1A), streptomycin (STR), and ethionamide (ETH). Although genes along with their 1 kbp upstream and downstream regions were used for primer design with oli2go, some resulting primers only partially covered the target genes (Fig. 1A). Initially, we used primers designed by oligo2go to test PCR reactions. However, for two long amplicons, rpoB and embB, gel electrophoresis results showed no amplified products. To address this issue, we divided these two genes into smaller segments for amplification. Through multiple trials and optimizations, we successfully resolved the issue of absent amplification products and non-specific bands, ultimately finalizing the optimized primer design combination.
A single 17-plex PCR assay with thermo-protection buffer for DNA preparation amplifies 16 resistance-associated genesAs shown in Fig. 1A, we targeted 16 genes associated with resistance to first- and second-line drugs, with expected PCR product lengths ranging from 1032 bp (rpsL) to 2698 bp (katG), as depicted in Fig. 1C. After rapid barcoding and nanopore sequencing, the average read lengths for the control (without) and test (with thermo-protection buffer) samples were 844.2 bp and 942.6 bp, respectively, while the mapped Mtb read counts significantly increased from 6183 to 32,301 reads. As depicted in Fig. 1C, the lowest mapped rate was observed for ahpC, with its rate increasing from 0.085 to 0.272%. While the mapped rate for each gene showed variability, the test sample exhibited a markedly higher overall percentage of mapped reads at 61.08%, compared to the control’s 5.73%. Additionally, the spike-in experiment (Fig. 1D) revealed that the overall mapped rate increased with higher concentrations of Mtb, and samples treated with the thermo-protection buffer exhibited greater fold changes in overall mapped rates than control samples. The highest peak was observed at an intermediated Mtb concentration (873 CFU/ml), with decent depth of coverage across 15 genes (Figure S1, barcode21). This appears to be the limit of detection for sequencing the 12 spike-in samples using a MinION flow cell. Consequently, this simple procedure utilizing the thermo-protection buffer dramatically enhances target sequencing performance.
Nanopore sequencing with rapid barcoding provides next-day antimicrobial resistance predictionWe initially applied the procedure to 12 clinical sputum samples (F01–F12, AFS 2+), all of which were susceptible to INH, RIF, EMB, and STR, using a Flongle flow cell. Although the Flongle ran for 24 h, we extracted 15-hour sequencing reads to simulate typical lab hours, ensuring next-morning results. As illustrated in Fig. 2, all samples except F02, F03, F07, and F09 exhibited at least three genes with coverage greater than 40, indicating the presence of Mtb. Detailed results are presented in Figure S2. AMR predictions were generated when more than eight genes had read coverage greater than 40. Therefore, an additional report for F11 was produced at the end of the Flongle run. Nevertheless, six samples were predicted correctly as susceptible using the Flongle. The total cost of $200 USD, including reagents and flow cell expenses, reduces the per-sample cost to less than $35. Subsequently, we used a MinION flow cell to analyze 40 clinical sputum samples, including 36 Mtb-positive (M01–M36) and 4 Mtb-negative (M37–M40). Despite of a 72-hour sequencing time, 15-hour reads were utilized to detect Mtb and predict AMR (Fig. 3, Figure S3). As depicted in the left panel of Fig. 3, Mtb was identified in 28 out of 36 Mtb-positive samples, with correct AMR predictions made for 21 of the 28 Mtb-detected samples. The prediction results obtained from the 17 clinical samples (M01–M12, and M20–M24) with high smear grades (AFS 2 + and above) demonstrated that our protocol is capable of providing accurate AMR predictions. Only one INH-resistant sample (M03) was falsely predicted as susceptible. As adequate sequencing reads were generated by MinION, we further extracted 4-hour reads to simulate sequencing conducted before noon, enabling results to be provided on the same day. In this scenario, correct AMR predictions were still obtained for the 13 clinical samples with AFS 2 + and above, plus one trace sample (M25, in the right panel of Fig. 3). Therefore, we recommend sequencing approximately 12 AFS 2 + and above samples at a time for optimal efficiency.
Fig. 2
Targeted gene profiling of twelve AFS 2 + sputum samples conducted using a Flongle flow cell, all showing phenotypic susceptibility to ethambutol (EMB), isoniazid (INH), rifampicin (RIF), and streptomycin (STR). Mycobacterium tuberculosis presence was determined by detecting reads from at least three genes with a coverage of more than 40 reads; otherwise, inconclusive presence was inferred. Furthermore, antimicrobial resistance (AMR) predictions were generated when more than eight genes had read coverage greater than 40. Bold font in AMR predictions indicates consistent predictions with pDST
Fig. 3
Targeted gene profiling of 40 sputum samples conducted with a MinION flow cell. This run included 36 Mtb-positive samples (M01-M36) and 4 Mtb-negative samples (M37-M40). Mycobacterium tuberculosis presence was determined based on the detection of reads from at least three genes with a coverage of more than 40 reads; otherwise, inconclusive presence was inferred. Furthermore, antimicrobial resistance (AMR) predictions were generated when more than eight genes had read coverage greater than 40. Bold font in AMR predictions indicates consistent predictions with pDST
Analysis of Mtb genomic sequences using 16 resistance-associated genes provides accurate AMR predictionsDue to the limited prevalence of drug-resistant strains in clinical samples, we employed our analytical pipeline to analyze publicly available nanopore genomic data and compared the prediction outcomes with Mykrobe and TBProfiler. As shown in Table 1 and Supplementary Table S2, out of 1417 AMR prediction for PRJNA650381, we found a 95.8% agreement between our ARapidTb predictions and pDST results, outperforming the other two WGS-based tools. Except for the low sample sizes (less than 5) in amikacin (AMK)-resistant and fluoroquinolone-resistant (i.e. ofloxacin (OFX)-resistant in Table 1) samples, we noted low sensitivities in predictions for pyrazinamide (PZA) and ethionamide (ETH). However, the low sensitivity for PZA was attributed to the high number of rare variants in the pncA gene [22]. Compared to Mykrobe and TBProfiler (Supplementary Table S2), the high number of false negatives generated by our method was attributed to deletion events in ethA, most of which were not included in Tbresdb. However, the known issue regarding indel characterization of nanopore sequencing resulted in high false positives for isoniazid and ethionamide predicted by Mykrobe and TBProfiler. Our ARapidTb results for PRJEB49093 (Table 2 and Supplementary Table S3) corroborated the findings of Hall et al. [20], confirming that genotyping resistance variants with nanopore sequencing was highly concordant with Illumina sequencing. Among the 1812 predictions made by Mykrobe, there was only one false positive and 30 false negatives. Although ARapidTb reduced the number of false positives to 21, it increased the number of false negatives to 15, mainly due to the omission of the gid gene in the primer design for predicting streptomycin (STR) resistance. Overall, our results from the nanopore WGS of 442 Mtb isolates show that our amplicon-based analysis tool, ARapidTb, is comparable to whole-genome analysis using Mykrobe and outperforms TBProfiler.
Table 1 Correlation between antimicrobial resistance predictions from WGS data of PRJNA650381 and drug susceptibility test (DST) results when availableTable 2 Correlation between antimicrobial resistance predictions from nanopore WGS data using various tools and Illumina WGS predictions using mykrobe for PRJEB49093
Comments (0)