
Remember me
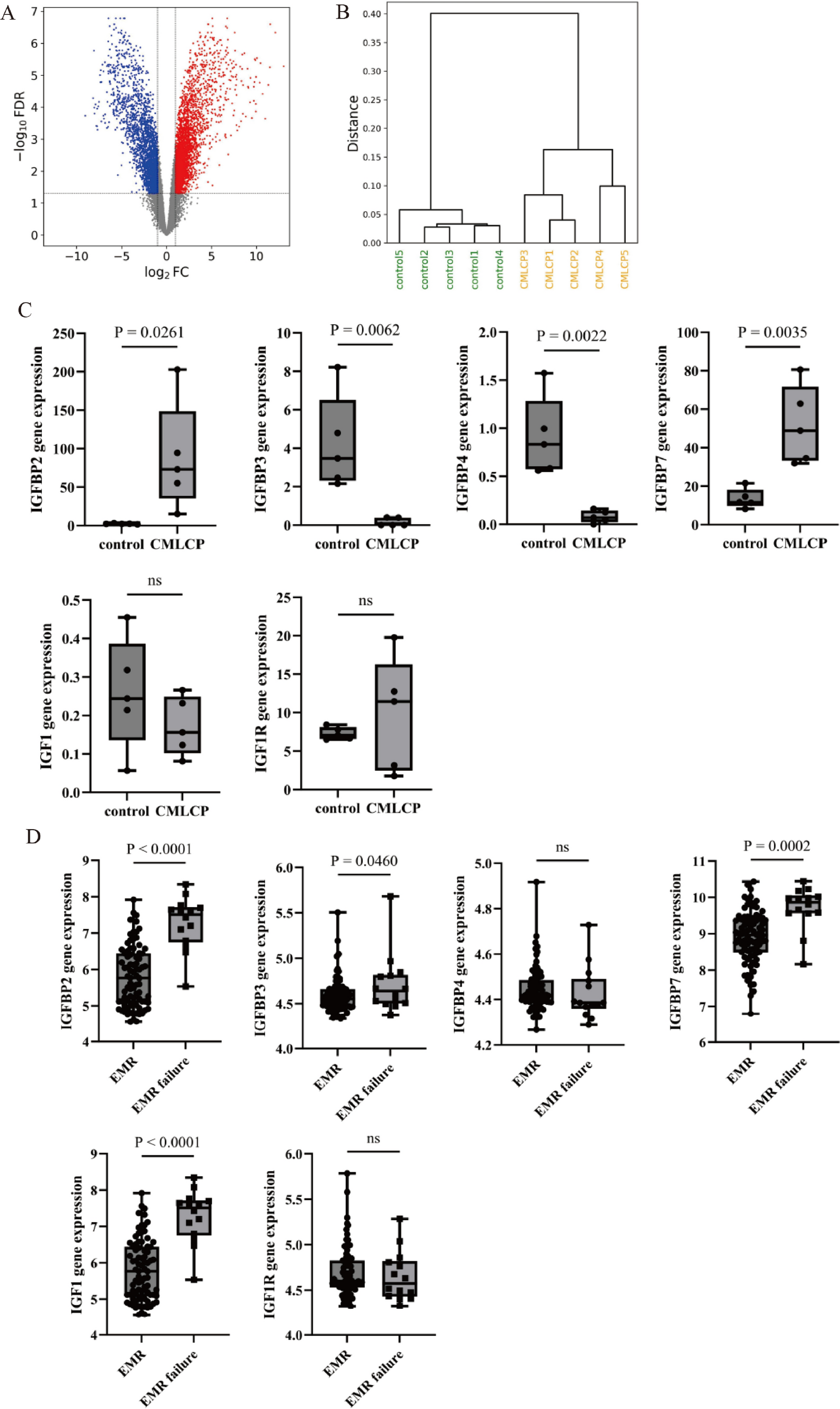
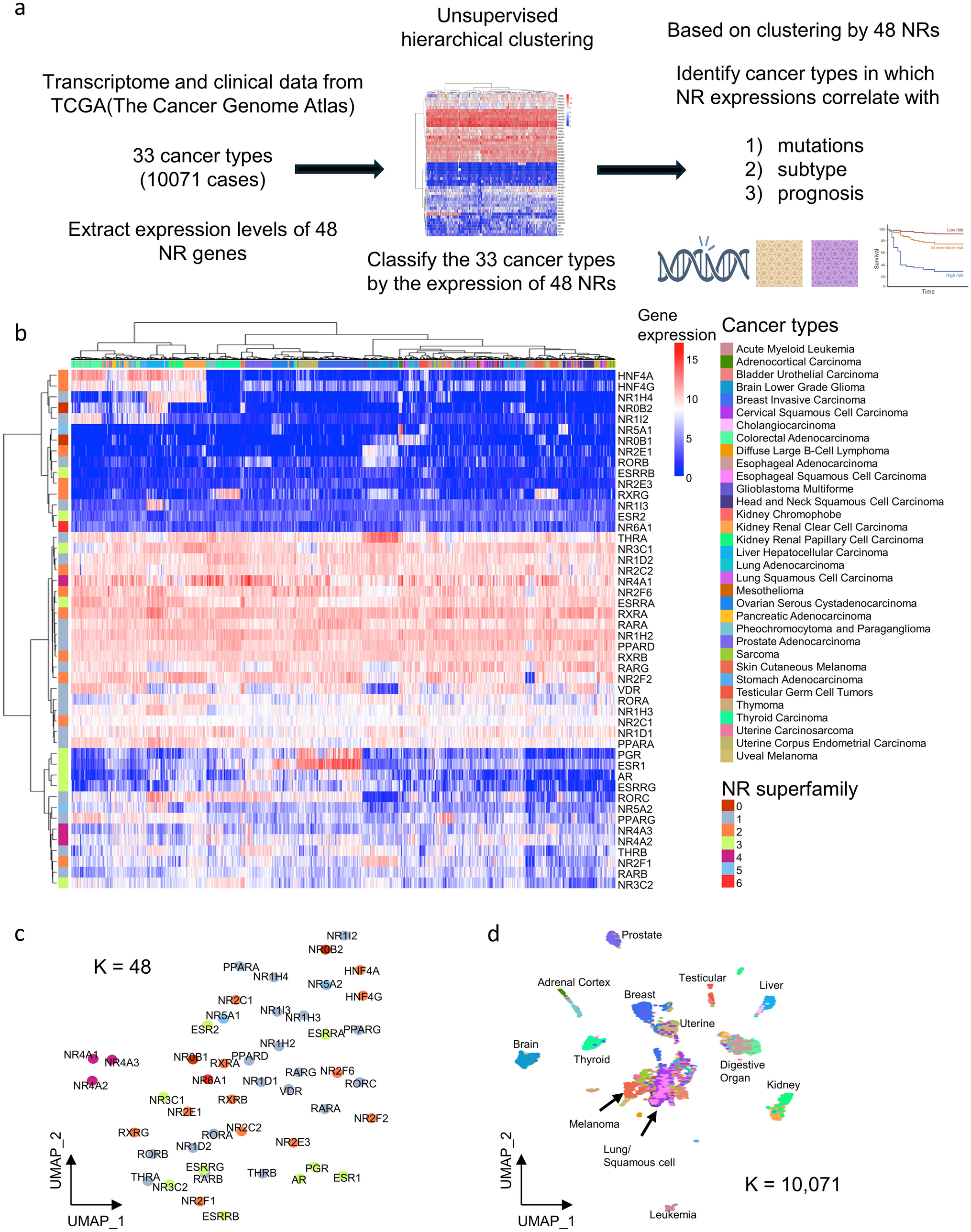
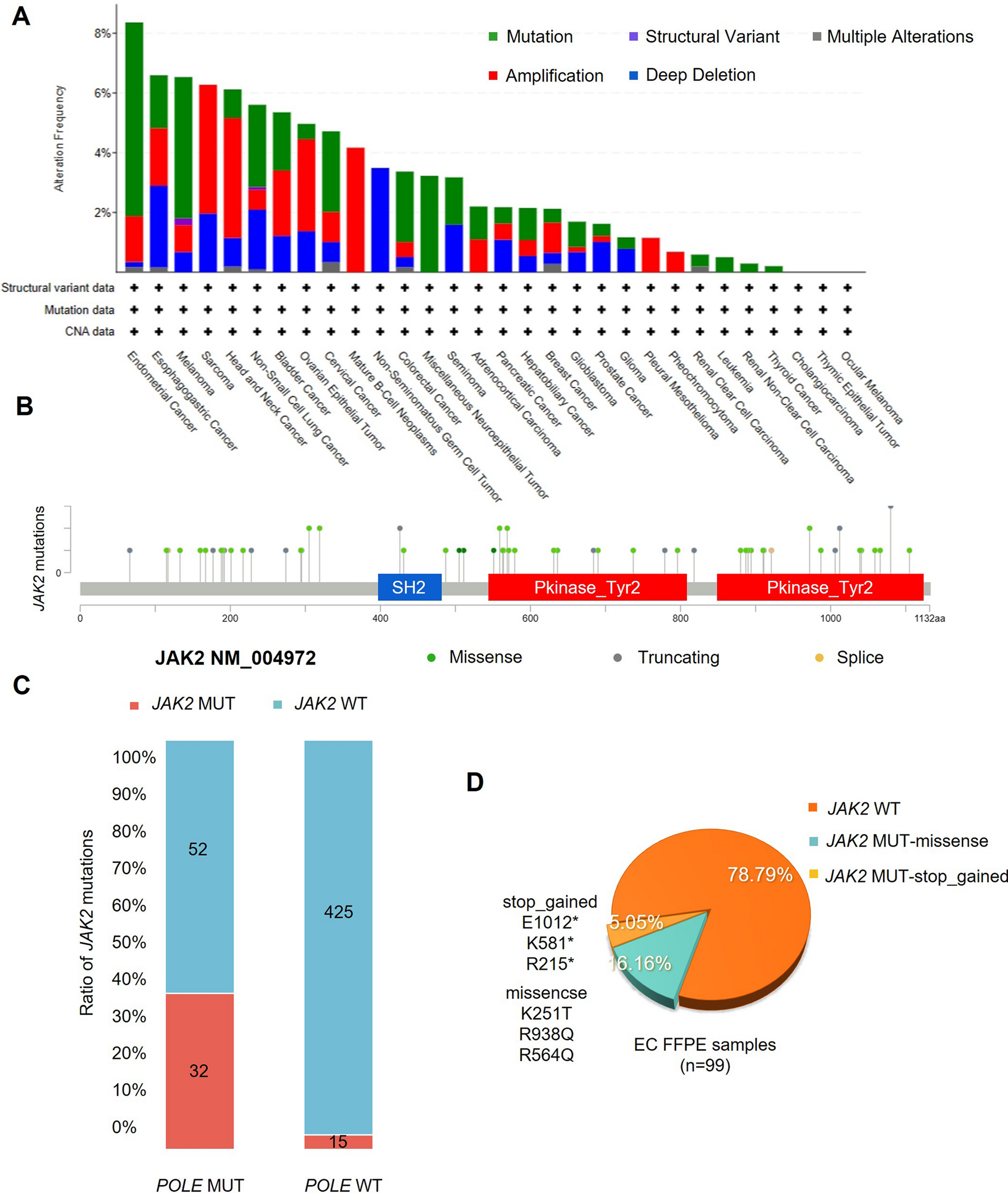
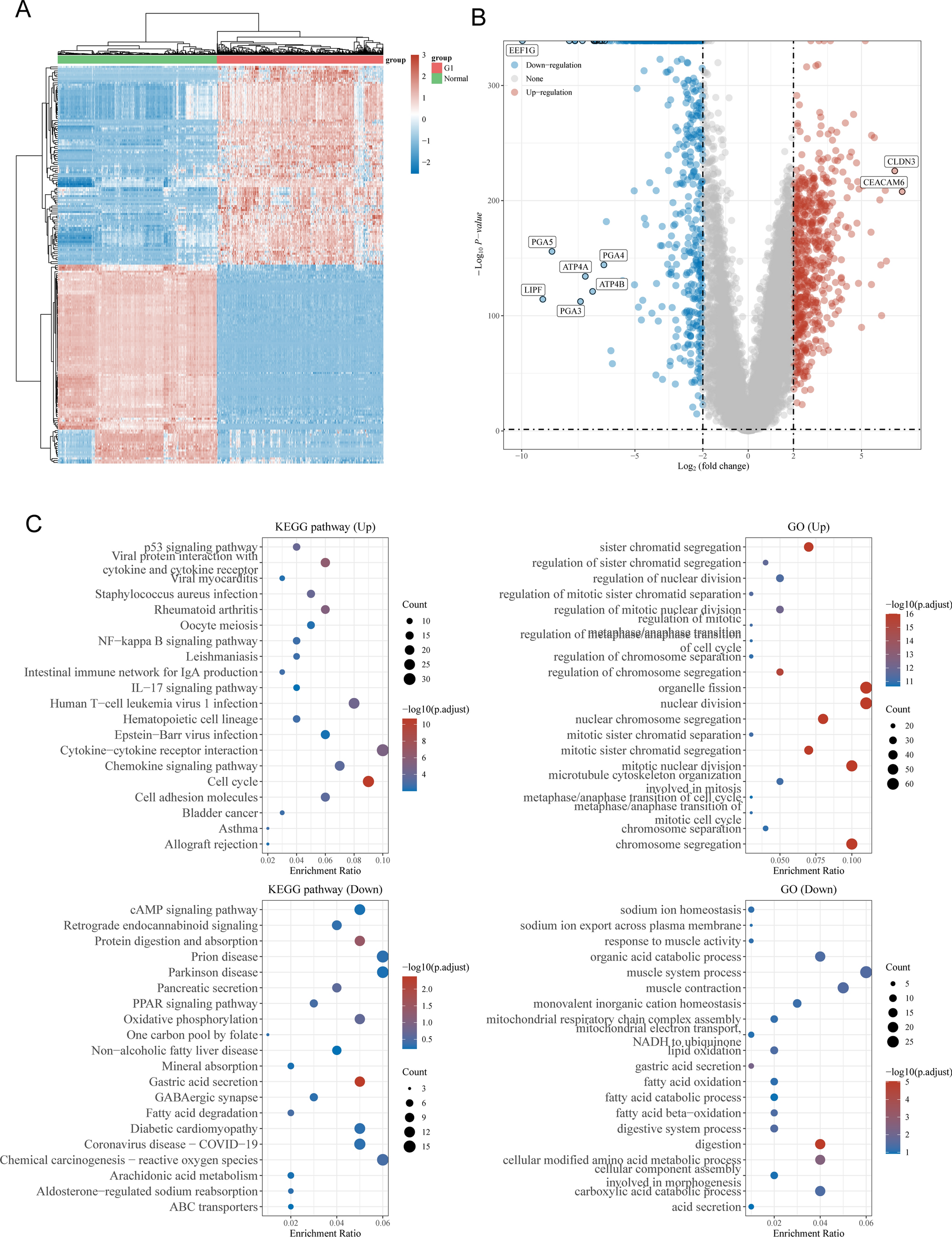
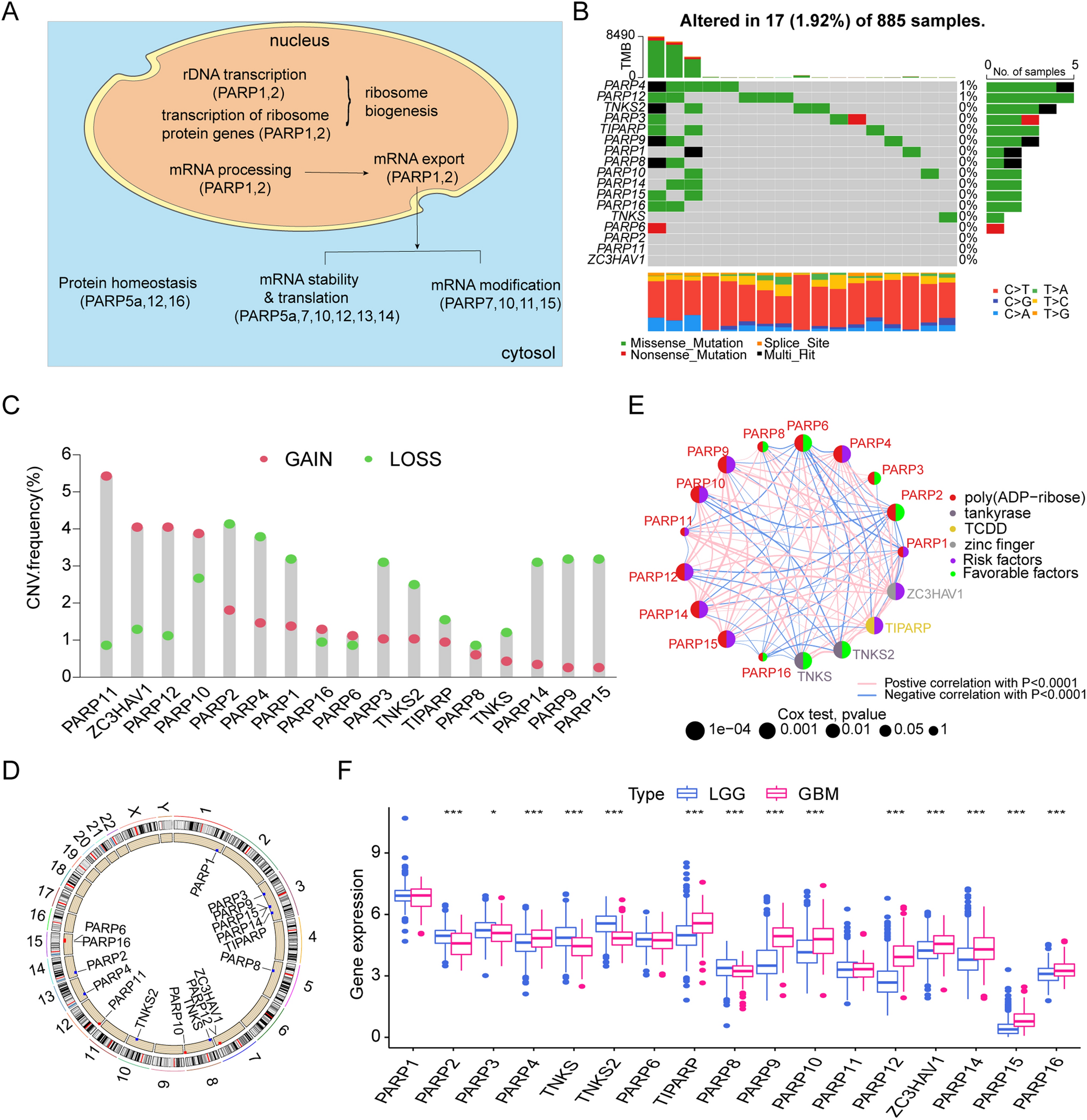

Figure 1A showed the chromosomal locations of a total of 44 glutamine metabolism related genes (GMRGs) from the REACTOME and GO databases. Based on their transcripts per million (TPM) values, their HRs were calculated between high and low expression groups as determined by optimal cut-off value (Fig. 1B). 24 of them (54.5%) were determined as risky factors. Compared with normal tissue in the TCGA-LUAD cohort, 26 out of 44 GMRGs had elevated expression level in tumor (Fig. 1C). Notably, the genetic alternations of GMRGs were observed in 184 out of 234 (78.63%) LUAD cases in the TCGA-LUAD cohort, and CPS1 mutation, mainly missense mutation, occurred in 27 per cent of all cases (Fig. 1D). Moreover, the CNV mutations of GMRGs were common, with most genes mutated with most genes mutated in nearly 50% of cases, and heterozygous amplification and deletion comprising the majority of the mutations (Fig. 1E).
Fig. 1
The expression, clinical correlation and mutation landscape of glutamine metabolism related genes (GMRG). A The chromosomal locations of 44 GMRGs. B The hazard ratios of GMRGs compared between a given high and low GMRG groups with optimal survival cut-off. C The expression level of GMRGs compared between tumor and normal tissues. The Wilcoxon rank-sum test was used to assess statistical significance. D The mutational landscape and E copy number variation mutation landscape of GMRGs
3.2 Consensus clustering with combination of GMRGs and CIBERSORTx resultsGlutamine metabolism has been shown to predominantly affect immunity, by serving as an essential nutrient for the proliferation of lymphocytes. As a result, we combined the expression GMRGs and the CIBERSORTx results in absolute mode, representing the absolute infiltration status of immune cells, to cluster the LUAD samples. The optimal k value of 3 was determined by the least proportion of ambiguous clustering (PAC) (Fig. 2A), and the clustering result was presented in Fig. 2B. There were significant survival differences among three clusters, and cluster 3 suffered from worst survival compared with the other two groups (all p value < 0.01) respectively (Fig. 2C). The summary of the clustering results, along with clinical information of patients was illustrated in Fig. 2D. Of note, cluster 1 and cluster 3 showed more activated glutamine metabolism, but with more infiltrated immune cells. With further analysis of clinical status distribution, there were larger proportion of patients with advanced stages (p = 0.084), T stages (p = 0.065), N stages (p = 0.032) in cluster 1 and cluster 3, and cluster 3 had more male patients (p = 0.001) (Fig. 2E). Given the worst survival in cluster 3, we filtered the elevated genes and immune cell infiltration in cluster 3. And a total of 11 GMRGs were upregulated in cluster 3 as compared with cluster 1 and cluster 2, of which 8 were risky factors (Fig. 2F, 1B).
Fig. 2
Consensus clustering with GMRGs and CIBERSORTx results. A The proportion of ambiguous clustering. B The consensus clustering results. C The KM plot of three GM clusters. The Log-rank Test was used to assess statistical significance. D The summary of clinical information, GMRG expression and CIBERSORTx value among three GM clusters. E The comparison of proportion of clinical indicators among three GM clusters. The Chi-square test was used to assess statistical significance. F The differential expression of GMRGs and CIBERSORTx value of GM cluster 3 compared with cluster 1 and cluster 2. The Wilcoxon rank-sum test was used to assess statistical significance
3.3 Establishment of the glutamine metabolism risk scoreThere were 42 GMRGs also detected by the GPL570 platform (excluding ARHGAP11B and GLYATL1B), which were then fitted into 101 machine learning algorithms (see Supplementary materials). The random survival forest model (RSF) demonstrated peak performances with highest average C-index (average C-index = 0.689) among datasets (Fig. 3A). And Fig. 3B showed the top 30 important variables in the RSF. In the integrated meta-LUAD cohort containing 1106 samples, the KM analysis indicated worse overall survival (OS) in the high-risk group using the median risk score as the cut-off point (Fig. 3C). Also with medium cut-off value, high-risk group had significantly worse OS in the TCGA-LUAD and GSE31210 cohort independently (Fig. 3D, E). And with optimal cut-off value, the high-risk group had significantly shorter OS (Fig. 3F, G). These results indicated that a RSF model built with GMRGs could predict effectively the OS of LUAD patients.
Fig. 3
Construction of the GM risk model. A Comparison of C-index among 101 machine learning models in 4 LUAD datasets. B The top 30 important variables in the RSF model. C–G The cut-off risk score for each dataset (top). The KM analysis results of LUAD samples (bottom). The Log-rank Test was used to assess statistical significance
3.4 Construction of a GMRG risk score-based nomogram and verification of its performanceThrough univariable Cox regression analysis, the GMRG risk score, male gender, advanced stages, patient age and history of smoking were risky factors (Fig. 4A). In the subsequent multivariable Cox regression analysis, the risk score, advanced stages, age and smoking history were independent risky factors (Fig. 4B). Considering all these risky factors,1 we established a survival nomogram (Fig. 4C). To build the nomogram with our clustering results, we evaluated the distribution of nomogram score in different GM clusters, and discovered that patients in the cluster 3 had higher nomogram score, indicating greater risk (Fig. 4D). Moreover, the calibration curve demonstrated that the survival nomogram score had high consistency between predicted OS and actual OS (Fig. 4E). The predictive performance of 1-, 3- and 5-year survival were evaluated using ROC curves, and the nomogram's prediction of OS had an area under the curve (AUC) of 0.874 at 1 year, 0.855 at 3 year and 0.806 at 5-year survival (Fig. 4F). And the time-dependent AUC value indicated a risk score-based GM nomogram had stable predicting value over time (Fig. 4G). The decision curve analysis (DCA) result showed that nomogram had more benefit for clinical decision compared with other indicators (Fig. 4H).
Fig. 4
Validation of the efficiency of GM risk model and construction of GM risk score-based nomogram. A The univariable Cox regression and B multivariable Cox regression analyses of GM risk score together with other clinical indicators. C Construction of GM risk score-based nomogram. D The distribution of nomogram score in the TCGA-LUAD cohort in different GM clusters. E Calibration curve showed the correlation between nomogram predicted OS and actual OS at 1-, 3-, and 5-year OS. F The ROC results of nomogram in the meta-LUAD cohort. G The time-dependent AUC value of nomogram score. H The DCA results of nomogram compared with other indicators
3.5 Cluster 3 LUAD patients had higher risk scores and aberrant cell proliferative activityAmong three GM clusters, cluster 3 exhibited the highest average risk scores, demonstrating a consistent relationship between GM molecular clusters and GM risk score (Fig. 5A). Glutamine metabolism was essential in the activation and maintenance of anti-tumor immunity. As was illustrated in Fig. 5B, samples in the cluster 3 were mainly risk high and immune cold, which may contribute to the poorest OS (Fig. 5B). When comparing cluster 3 samples with the other two clusters, a total of 131 up-regulated and 102 down-regulated genes were obtained using limma (Fig. 5C). These down-regulated genes were correlated with Gene ontology (GO) biological processes (BP) including positive regulation of T cell mediated cytotoxicity and antigen processing and presentation, highlighting the immune cold landscape in cluster 3 (Fig. 5D). While these up-regulated genes were enriched in cell proliferation related GO BPs, including cell division, mitotic cell cycle, mitotic spindle assembly checkpoint, indicating aberrant proliferative activity, and was located in the centrosome, nucleoplasm, cytosol and nucleus (Fig. 5E). In addition, the GSEA results showed that the top variable genes were enriched in Hallmark G2M checkpoint, E2F targets and mitotic spindle, further validating the GO results (Fig. 5F).
Fig. 5
GM cluster 3 samples had highest average risk score and aberrant proliferating activity. A Comparison of GM risk score among three GM clusters. The Wilcoxon rank-sum test was used to assess statistical significance. (***: p < 0.001) B Sankey plot showed the connection of GM clusters, GM risk score and Immune score by ESTIMATE. C The differentially expressed genes (DEGs) of GM cluster 3. D, E The gene ontology (GO) results of biological process (BP) and cellular component (CC) of cluster 3 DEGs. F The GSEA results of DEGs
3.6 Single-cell RNA sequencing data determined 4 GMRGs as hub genes in LUADBased our previous identification of genes that were risky factors, up-regulated in cluster 3 and were included in the establishment of RSF, a total of 7 overlapping genes were finally filtered (Fig. 6A). To validate the expression level of them in cancer cells, a single-cell RNA sequencing data including 11 samples (Fig. 6B). According to canonical cell markers, these cells were annotated into 8 major cell types (Fig. 6C, D). And compared with the annotation by Kim N et al., the annotated epithelial cells were highly consistent[16] (Fig. 6E). In each patient, the relative expression level of PYCR1, ALDH18A1, SLC38A2 and RIMKLB was relatively abundant (Fig. 6F). And the umap plot illustrated the absolute expression of 4 GMRGs among cell types (Fig. 6G).
Fig. 6
Single-cell RNA data validated the GM hub genes in LUAD cancer cells. A The overlap of risky factors, up-regulated genes in cluster 3 and genes used in the RSF model. B Umap plot of the summary of the integration results of 11 LUAD samples. Boxplot showed the number of cells in each sample after quality control. (C) The expression patterns of canonical cell markers among cell clusters. D The annotated major cell types as referred to cluster markers. E Comparison of our annotation with the annotation by Kim N et al. F The relative expression of 7 GM hub genes among patients. G The expression level of 7 GM hub genes in different cell types
3.7 ALDH18A1 exhibited the strongest correlation with patient overall survivalALDH18A1 exhibited an AUC value greater than 0.55 across all four LUAD datasets for both 1-year and 3-year survival (Fig. 7A, B). In the external CPTAC-LUAD database with FPKM-format RNA sequencing data, higher expression of ALDH18A1, SLC38A2, and RIMKLB correlated with shorter overall survival (OS) (Fig. 7C). Based on the results presented above, ALDH18A1 exhibited the strongest correlation with OS in LUAD patients. The expression level of ALDH18A1 was significantly higher in stage II compared to stage I, in advanced T stages compared to T1, and in N2 compared to N0 (Fig. 7D). At the protein level, ALDH18A1 was higher in tumor tissue compared to normal tissue (Fig. 7E). The average protein level increased with advanced stages and was significantly higher in stage III compared to stage I (Fig. 7F). Furthermore, higher protein levels of ALDH18A1 were associated with shorter OS in the CPTAC-LUAD cohort (Fig. 7G).
Fig. 7
ALDH18A1 exhibited the strongest correlation with patient overall survival. A, B The ROC analysis results of four candidate hub genes at A 1-year OS and B 3-year OS in 4 LUAD datasets. C The KM analysis of ALDH18A1, PYCR1, SLC38A2 and RIMKLB at mRNA level in the CPTAC-LUAD cohort. D The correlation of ALDH18A1 with clinical stages, T, N and M. E The protein level of ALDH18A1 compared between tumor and normal tissues in the CPTAC cohort. F The correlation of ALDH18A1 protein level with clinical stages. The Wilcoxon rank-sum test was used to assess statistical significance. G The KM analysis of ALDH18A1 at protein level in the CPTAC-LUAD cohort. The Log-rank Test was used to assess statistical significance
3.8 The up-regulated expression of ALDH18A1 was correlated with cell proliferation and immune exclusionA total of 76 upregulated genes were filtered in the high ALDH18A1 expression group (Fig. 8A), and genes were enriched in pathways involved in the formation of the extracellular environment, such as BPs like blood coagulation, fibrin clot formation, and cell–matrix adhesion; CCs including the fibrinogen complex and platelet alpha granules; and MFs such as extracellular matrix structural constituent (Fig. 8B). Moreover, levels of T cells CD8, T cells follicular helper, Macrophages M1 and M2 were significantly higher in the low ALDH18A1 subgroup, as well as higher stromal score, immune score and ESTIMATE score (Fig. 8C, D). And the expression of ALDH18A1 were tightly negatively correlated with immune score, and numerous immune cells (Fig. 8E). The results suggested that ALDH18A1 may contribute to the establishment of an immune-excluded extracellular environment, which finally led to an immune-cold tumor microenvironment and ultimately resulted in shorter OS.
Fig. 8
High ALDH18A1 subgroup had abnormal proliferating activity and immune cold tumor microenvironment. A The DEGs between high and low ALDH18A1 subgroups. B The GO results of up-regulated DEGs in high ALDH18A1 subgroup. C, D Comparison of C CIBERSORTx results and D ESTIMATE results between two ALDH18A1 subgroups. The Wilcoxon rank-sum test was used to assess statistical significance. E The Spearman correlation results of ALDH18A1 value with CIBERSORTx and ESTIMATE results
Comments (0)