2.1 Public data download
Single-cell RNA sequencing (scRNA-seq) data related to ovarian cancer were obtained from the gene expression omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). The dataset GSE217517 encompasses ovarian cancer tumor tissues from 8 ovarian cancer patients (GSM6720925-GSM6720932). The data was analyzed using the R software package "Seurat" [24]. Data quality control was performed based on the criteria of 200 < nFeature_RNA < 5000 percent.mt < 20, and highly variable genes expressing the top 2000 variances were screened. As the data were obtained from a public database, no ethical approval or informed consent was required [25].
2.2 Cluster analysis
To reduce the dimensionality of the scRNA-Seq dataset, principal component analysis (PCA) was performed based on the top 2000 highly variable genes by variance. The top 20 principal components (PCs) were selected for downstream analysis using the Elbowplot function in the Seurat software package. The FindClusters function provided by Seurat was employed to identify main cell subgroups, with the resolution set at the default value (res = 1). Subsequently, the UMAP algorithm was utilized to reduce the nonlinear dimensionality of the scRNA-seq sequencing data. Markers for various cell subgroups were identified using the Seurat software package. The cells were annotated by combining known cell lineage-specific marker genes with manual annotations using the CellMarker online tool in conjunction with the "Singel R" package [26]. Cell communication analysis was performed using the "CellChat" package in the R programming language.
2.3 GO and KEGG enrichment analysis
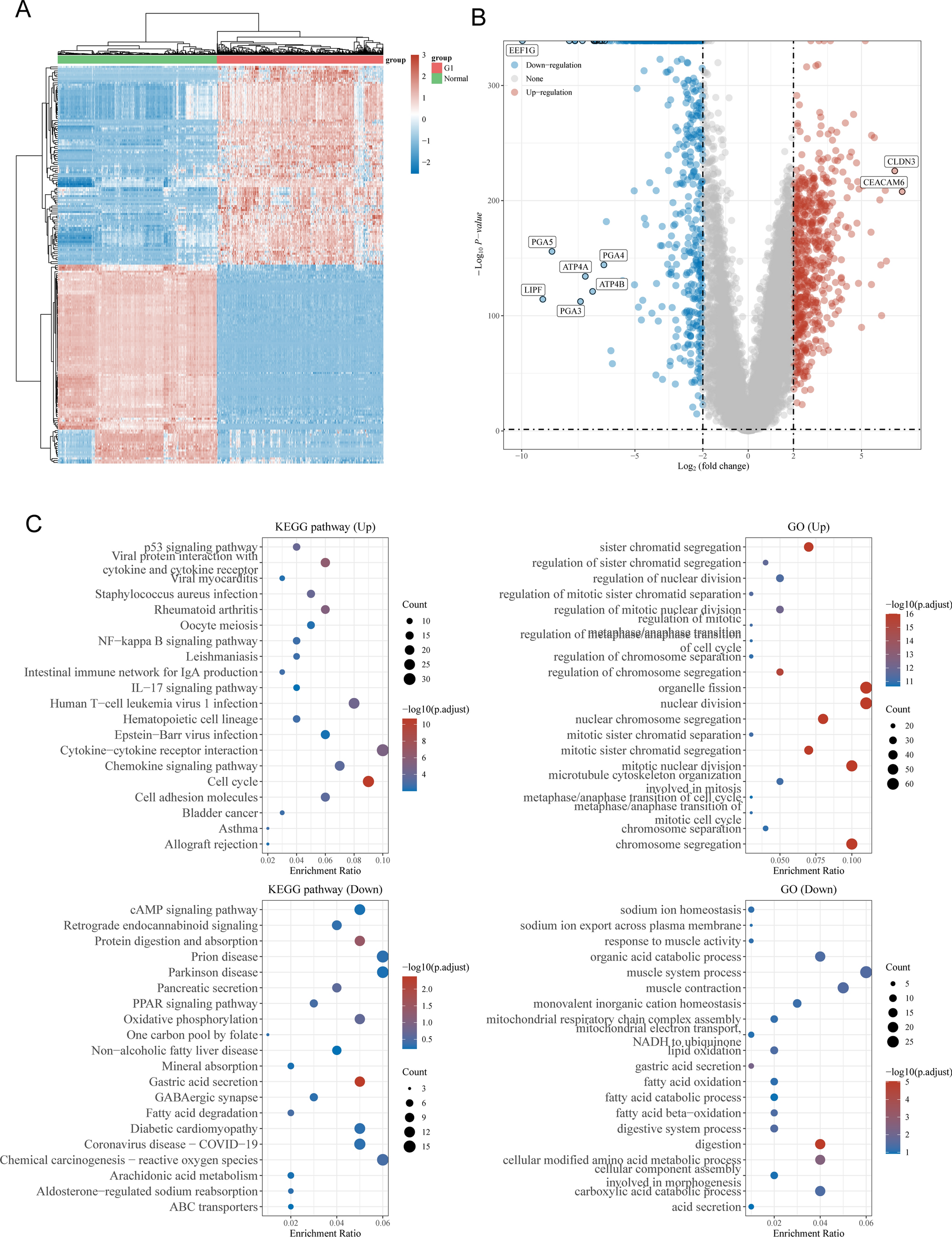
After clustering, the differential feature genes in each cell type were analyzed using the FindAllMarkers function. Specifically, the characteristic genes of epithelial cells or significantly differentially expressed genes (DEGs) from high-throughput transcriptomic data were selected. Subsequently, the co-expressed genes were subjected to gene ontology (GO) functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis using the clusterProfiler package in R software. The results were visualized using the ggplot2 package [27].
2.4 High-throughput transcriptome sequencing
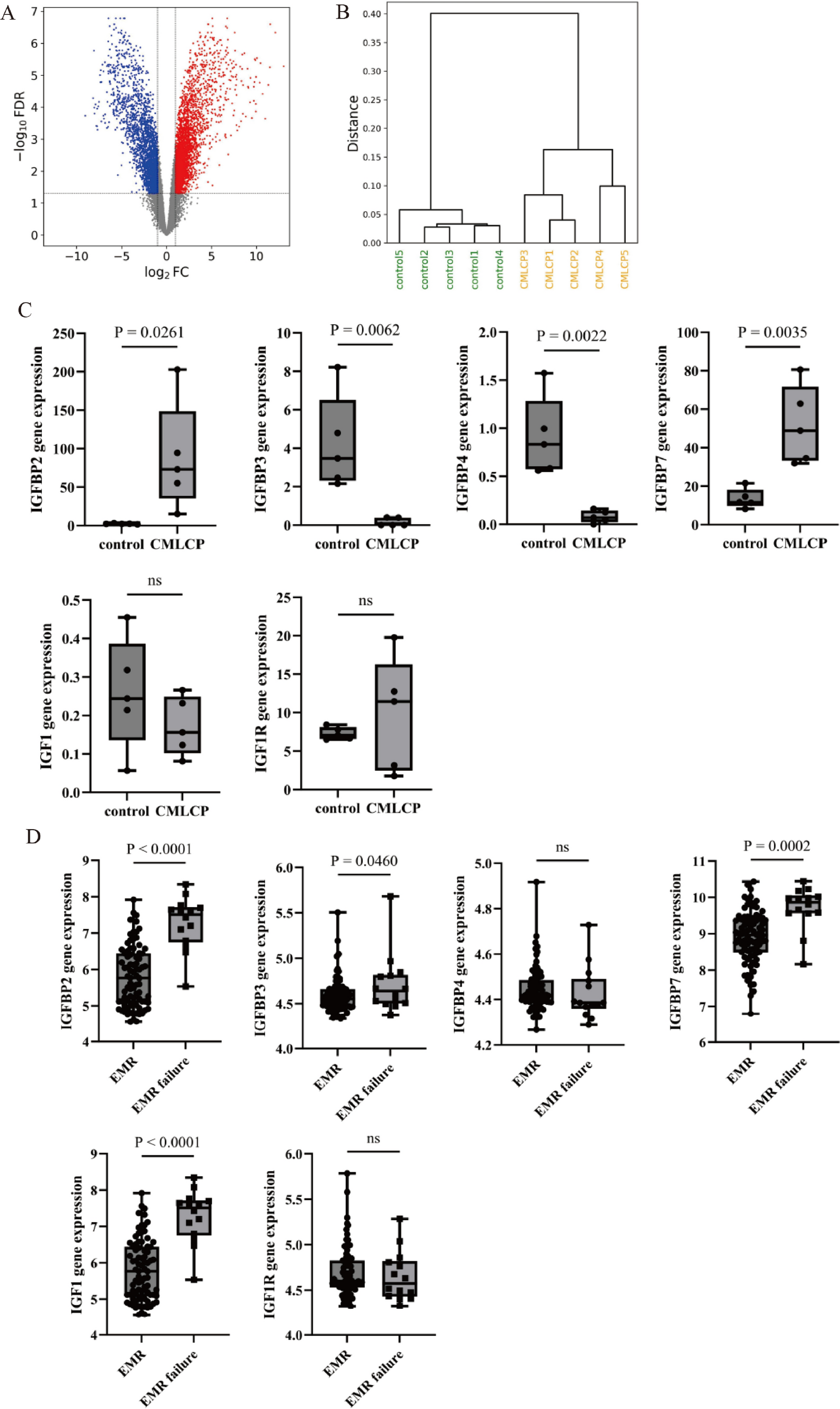
SK-OV-3 ovarian cancer cells treated with GEM (cultured in RPMI-1640 medium containing 1 μM GEM at 37 °C for 5 days) and untreated SK-OV-3 ovarian cancer cells were collected, with three replicates for each group. A total of at least 1 μg of total RNA was isolated and purified using DNAse I and a silica membrane (RNeasy Kit, 74,004, Qiagen, Hilden, Germany). RNA was quantified using the Qubit RNA HS Assay Kit (Q32852, Thermo Fisher, USA) and diluted to 100 ng/μL. The quality of total RNA was confirmed using a Fragment Analyzer (Agilent 5400, USA). Samples with RNA quality ≥ 6 were selected for RNA library preparation in accordance with ISO/IEC-17025 certification (TruSeq RNA Library Preparation Kit v2, RS-122-2302, Illumina, USA). mRNA was enriched and fragmented using oligo(dT) magnetic beads (MS04T, Suzhou Walden Biotechnology Co., Ltd., Suzhou, China), followed by cDNA synthesis, adapter ligation, and PCR amplification. Paired-end sequencing with read lengths of 126 bp was performed on the Illumina HiSeq 2500 V4 sequencer (Illumina, USA), with each sample generating at least 12.5 Gbp, yielding approximately 50 million read pairs, according to the manufacturer’s protocol. Image analysis, base calling, and quality control were carried out using the Illumina data analysis pipeline RTA v1.18.64 and Bcl2fastq v1.8.4. RNAseq reads were provided in compressed Sanger FASTQ format [28]. DEGs were identified from the RNA-Seq dataset using the "edgeR" package in R software, with |logFC|> 2 and P.adjust < 0.05 as the screening criteria.
2.5 Analysis of protein–protein interaction (PPI) network
The STRING database (https://string-db.org/) was utilized to annotate the PPI network of DEGs. The data was exported as a TSV file, and genes correlated to candidate genes were selected. Subsequently, visualization was performed using Cytoscape software [29].
2.6 Analysis of gene expression in TCGA database
Data for ovarian serous cystadenocarcinoma from TCGA and corresponding normal tissue data from GTEx were retrieved from the Xiantao Academic website (TCGA_GTEx-OV). The normal control group included 88 samples, while the tumor tissue group included 427 samples. Statistical analysis was conducted using the Wilcoxon rank sum test, and data visualization was carried out using the ggplot2 package [30].
2.7 Correlation analysis
Pairwise correlation analysis was performed on the selected candidate genes from the high-throughput transcriptome data using Spearman correlation analysis. The results were visualized using a heatmap, with correlation coefficients ranging between -1 and 1, indicating the strength and direction of the correlation. The magnitude of the coefficient signifies the degree of correlation [31].
2.8 Cell culture and grouping
The human ovarian surface epithelial cells (IOSE-80) were purchased from Pricella (CP-H055). These primary ovarian epithelial cells were cultured in dishes coated with mouse tail collagen I (2–5 μg/cm2). The cells were cultured in RPMI1640 medium (Gibco, Cat. No: 11875119) containing 10% FBS (12483020, Gibco), 1% glutamine, and antibiotics (penicillin, 100 U/ml; streptomycin, 100 μg/ml) at 37 °C in a 5% CO2 humidified environment and could be passaged for 2–3 generations [32]. The human ovarian cancer cell line SK-OV-3 was obtained from the Cell Bank of the Chinese Academy of Sciences (TCHu185), and human umbilical vein endothelial cells (HUVECs, CRL-1730 ™ purchased from ATCC, USA) were cultured in RPMI1640 medium (Gibco, Cat. No: 11875119) containing 10% FBS, 1% glutamine, and antibiotics (penicillin, 100 U/ml; streptomycin, 100 μg/ml) at 37 °C in a 5% CO2 humidified environment and passaged twice a week [33].
GEM hydrochloride was purchased from MCE (LY 188011) and dissolved in sterile water. SK-OV-3 cells were cultured in RPMI-1640 medium (Gibco, Cat. No: 11875119) containing 1 μM GEM at 37 °C for 5 days, with one medium change during the treatment. This group was designated as the SK-OV-3/GEM group [34, 35]. This treatment was used to assess cell proliferation, migration, Transwell invasion, and angiogenesis.
Based on the known HIF-1α, VEGF-B and FGFR1 sequences in NCBI, Shanghai Hanheng Biotechnology Co., Ltd. (Shanghai, China) was commissioned to construct oe-NC, oe-HIF-1α, oe-VEGF-B and oe-FGFR1 into the lentiviral vector pHBLV-CMV-MCS-EF1-Puromycin. When SK-OV-3 cells were in the logarithmic growth phase, they were dissociated using trypsin and seeded at a density of 1 × 105 cells per well in 6-well plates. After routine incubation for 24 h, when the cell confluence reached around 75%, the medium containing an appropriate amount of packaged lentivirus (MOI = 10, working titer approximately 5 × 106 TU/mL) and 5 μg/mL polybrene (Merck, TR-1003, USA) was added for infection. The titer of lentivirus was 1 × 108 TU/ml.
Subsequently, after 72 h, the medium was replaced with a medium containing 4 μg/mL puromycin (Invitrogen, A1113803), and the cells were cultured for at least 14 days. Puromycin-resistant cells were expanded in a medium containing 2 μg/mL puromycin (Invitrogen, A1113803) for 9 days before being transferred to a puromycin-free medium for further cultivation, thereby obtaining SK-OV-3 cells stably overexpressing HIF-1α, VEGF-B or FGFR1[36].
The commercial FGFR1 silencing lentivirus (sc-39840-V) was purchased from Santa Cruz Biotechnology (Shanghai) and titrated to 109 TU/mL. After 72 h of infection, the medium was replaced with a medium containing 4 μg/mL puromycin, and the cells were cultured for at least 14 days. Puromycin-resistant cells were expanded in medium containing 2 μg/mL puromycin for 9 days, then transferred to a puromycin-free medium, resulting in a stable FGFR1-silenced cell line. SK-OV-3 cells with stable overexpression of HIF-1α were seeded at a density of 1 × 106 cells per well in a 6-well plate, and after 24 h of incubation, the cells were infected with lentivirus. The next experiment was conducted 72 h post-infection.
Cell groupings were as follows:
Group 1: (1) oe-NC (IOSE-80 cells infected with oe-NC lentivirus); (2) oe-HIF-1α (IOSE-80 cells with stable HIF-1α overexpression); (3) oe-VEGF-B (IOSE-80 cells with stable VEGF-B overexpression); (4) oe-FGFR1 (IOSE-80 cells with stable FGFR1 overexpression).
Group 2: (1) control group (SK-OV-3 cells); (2) GEM group (SK-OV-3 cells treated with GEM); (3) oe-NC + GEM group (oe-NC lentivirus-infected cells treated with GEM); (4) oe-HIF-1α + GEM group (cells with stable HIF-1α overexpression treated with GEM); (5) oe-HIF-1α + sh-FGFR1 + GEM group (cells with stable HIF-1α overexpression infected with sh-FGFR1 lentivirus and treated with GEM).
2.9 RT-qPCR analysis
All RNA samples were extracted from cells and tissues using a lysis buffer (Vazyme, R401-01). Complementary DNA (cDNA) was synthesized from mRNA samples (2 μg) using a reverse transcription system. Quantitative real-time PCR was performed using the QuantStudio™ 3 Real-Time PCR System (Applied Biosystems, Waltham, MA, USA) by mixing cDNA samples, target gene primers, and SYBR Green PCR Master Mix (Thermo Fisher Scientific Inc. 1176202K) for quantitative real-time PCR analysis [37]. The primer sequences used in this study are presented in Table S1 (primer designs sourced from Primer Bank).
2.10 Western blot (WB)
Cells from each group were collected and lysed on ice for 30 min using RIPA lysis buffer containing 1% PMSF (P0013B, Beyotime, Shanghai, China). After centrifugation at 14,000 g for 10 min at 4 °C, the supernatant was collected. Protein concentrations were determined using the BCA method (P0012S, Beyotime, Shanghai, China). An appropriate amount of 5 × loading buffer was added, and the samples were boiled at 100 °C for 10 min for protein denaturation. A total of 50 μg of protein was loaded per sample. The protein samples were separated by SDS-PAGE using a separating gel and a stacking gel. After electrophoresis, the protein bands were transferred onto a PVDF membrane. The membrane was blocked with 5% skim milk at room temperature for 1 h, followed by overnight incubation with primary antibodies at 4 °C. GAPDH was used as the loading control. After washing with phosphate-buffered saline with Tween (PBST), the membrane was incubated with HRP-conjugated goat anti-rabbit IgG secondary antibody at room temperature. Signal detection was then performed using the ECL detection system (32,209, Thermo Fisher Scientific, USA), and the membrane was exposed using an imaging system (Amersham Imager 600, USA) [38, 39]. Grayscale analysis was conducted using Image J. The experiment was repeated three times. Information on the antibodies used for Western Blot is provided in Table S2.
2.11 CCK-8 cell proliferation experiment
The cell proliferation assay was performed using the cell counting kit-8 (CCK-8) (Beyotime, C0037, Shanghai, China). Cells were seeded in a 96-well plate. At Day 0, 1, 2, 3, 4, and 5, 10 μL of CCK-8 solution was added to each well. Subsequently, the cells were incubated for 1 h, and the optical density of the cells was measured at 450 nm [40]. Each group was set up with 6 replicates, and the experiment was repeated thrice.
2.12 Wound healing assay
The migration of ovarian cancer cells was evaluated through a scratch closure assay. When cells reached 90% confluence in a 6-well plate, a straight line was drawn through the monolayer using a sterile 200 µL pipette tip. The floating cells were then washed off with PBS, and the original growth medium was replaced with a medium containing 1% fetal bovine serum to inhibit cell proliferation. Images of the scratch were captured at 0 and 24 h post-incision using an inverted microscope (Olympus, Japan), and the wound closure area was quantitatively analyzed using ImageJ software (NIH, USA) to estimate the cell migration ability [40].
2.13 Transwell invasion assay
The in vitro cell invasion assay was conducted using Transwell chambers (8 μm pore size; Corning, USA). Matrigel-coated Transwell chambers with 8 μm pores were prepared by adding 600 mL of FBS-containing medium to the lower chamber and incubating at 37 °C for 1 h. Digestion-released cells were resuspended in DMEM medium without FBS, and 2 × 104 cells/mL were seeded into the upper chamber. The chambers were then incubated at 37 °C with 5% CO2 for 24 h. Subsequently, the Transwell chambers were removed, washed twice with PBS, fixed with 5% glutaraldehyde at 4 °C, stained with 0.1% crystal violet for 5 min, rinsed with PBS, and any surface cells were removed with a cotton swab. Cell invasion was assessed under an inverted fluorescence microscope (Nikon TE2000, China), where 5 random fields were captured for each chamber, and the average number of cells passing through the chamber was calculated as representative of each group [41]. Each experiment was repeated thrice.
2.14 Angiogenesis assay
Angiogenesis was assessed using the Chemicon® In Vitro Angiogenesis Assay Kit (Chemicon International, ECM625) to evaluate blood vessel formation. Human umbilical vein endothelial cells (HUVECs) were utilized for the experiment following these procedures: ECMatrix™ solution was thawed overnight at 4 °C. 900 μL of the ECMatrix™ solution was mixed with 100 μL of 10 × ECMatrix™ Dilution Buffer in a sterile microcentrifuge tube. Then, 50 μL of the mixture was transferred to each well of a pre-chilled 96-well tissue culture plate and incubated at 37 °C for at least 1 h to allow the matrix solution to solidify. Endothelial cells were harvested and resuspended in the supernatants extracted from ovarian cancer cells of the control group, GEM group, oe-NC + GEM group, oe-HIF-1α + GEM group, and oe-HIF-1α + sh-FGFR1 + GEM group. Cells were seeded onto the polymerized surface of ECMatrix™ at a density of 1.0 × 106 cells per well and then incubated at 37 °C for 12 h. The formation of cellular networks was completed within 18 h, with initial signs visible after 4 h. The process of angiogenesis was observed using an inverted light microscope [42].
2.15 Statistical analysis
The data were obtained from at least three independent experiments, and the results are presented as mean ± standard deviation (Mean ± SD); for comparisons between the two groups, a two-sample independent t-test was employed; for comparisons involving three or more groups; one-way analysis of variance (ANOVA) was conducted. If the ANOVA results indicated significant differences, Tukey's Honestly Significant Difference (HSD) post-hoc test was performed for further comparisons between groups. For non-normally distributed or unequal variances data, the Mann–Whitney U test or Kruskal–Wallis H test was utilized. All statistical analyses were carried out using GraphPad Prism 9 (GraphPad Software, Inc.) and R language. The significance level for all tests was set at 0.05, with a two-tailed p-value less than 0.05 considered statistically significant.


Comments (0)