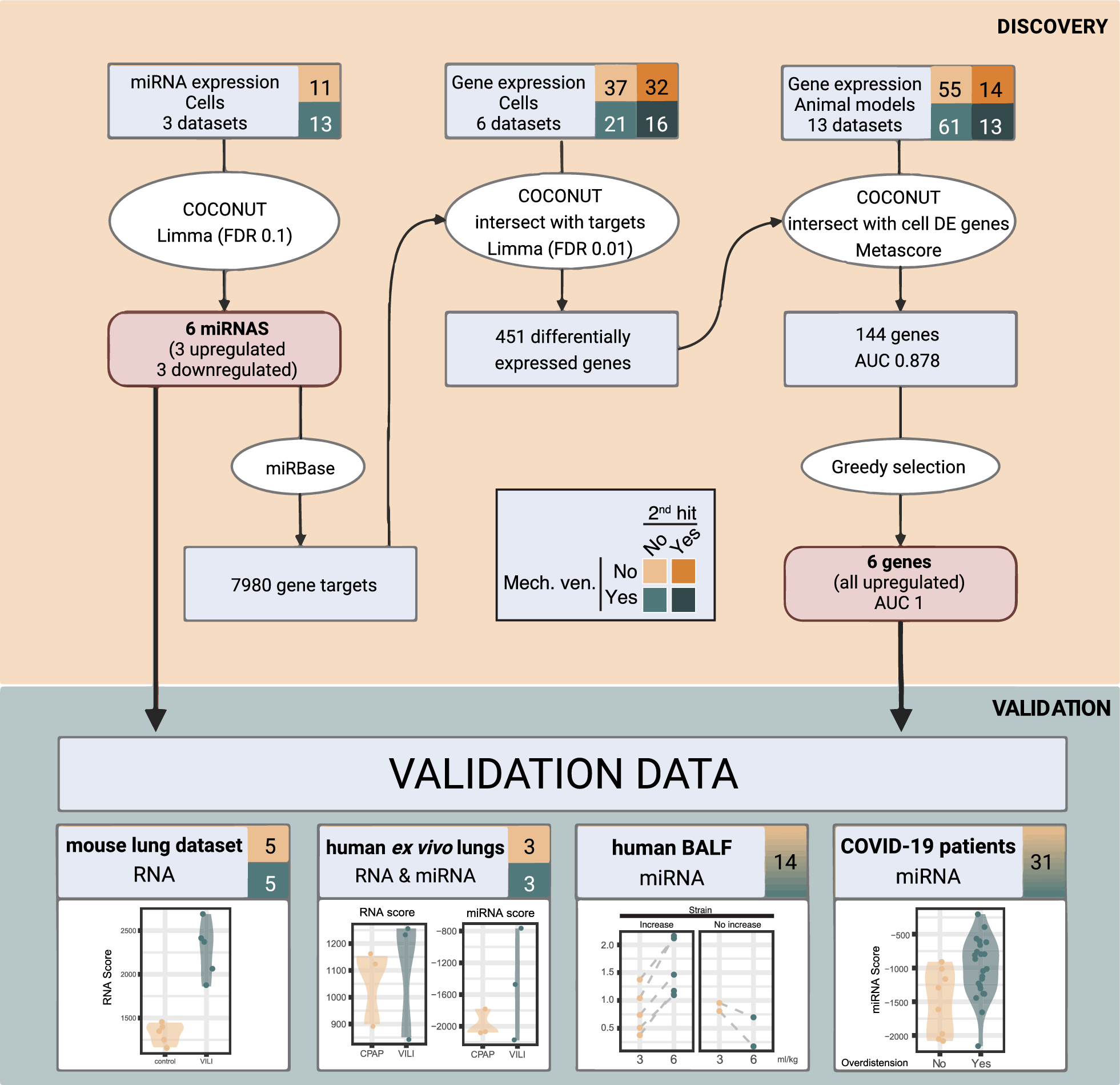
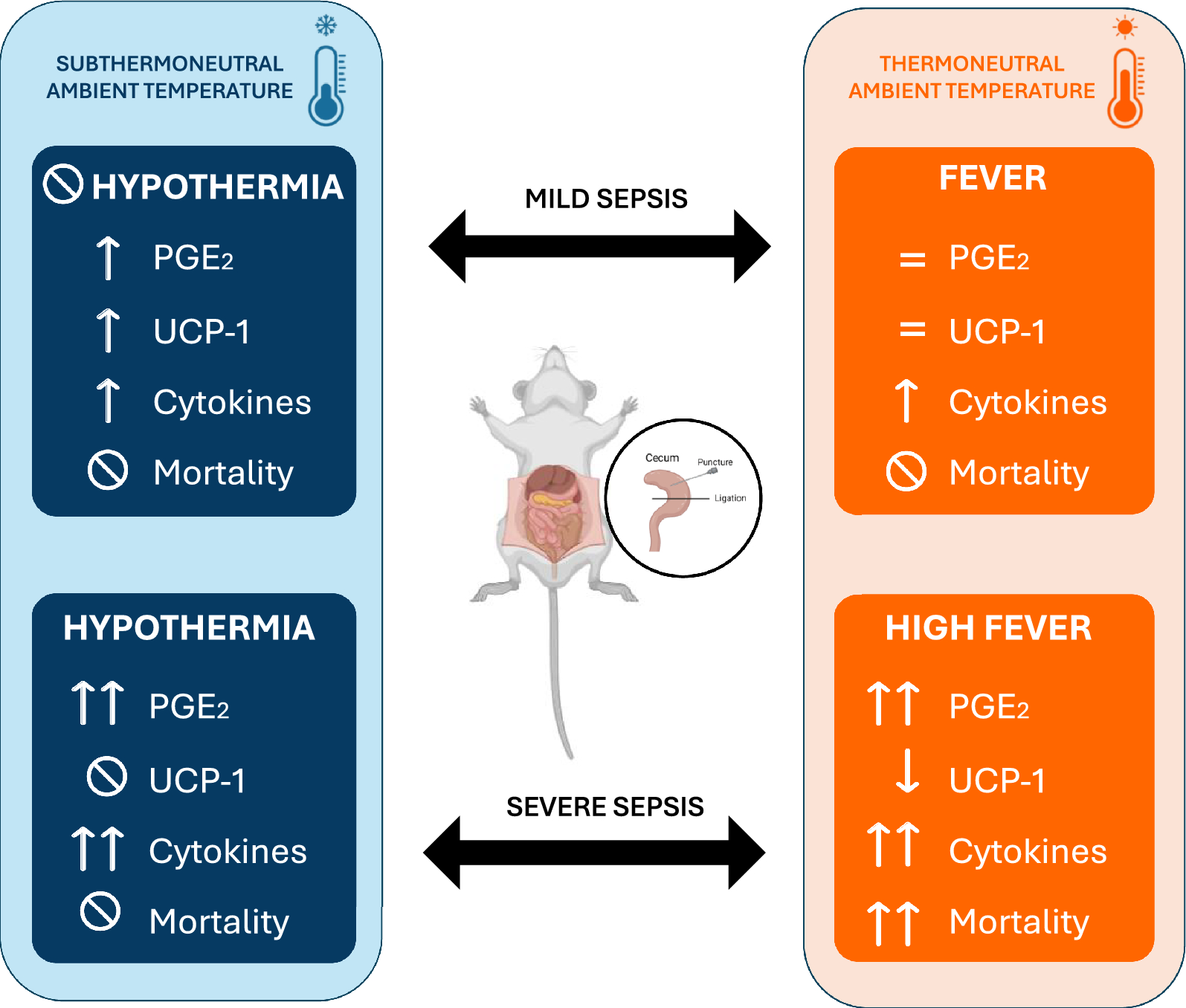
In this study, we investigated how both ambient temperature and infection severity affect body temperature in animals subjected to cecal ligation and puncture-induced sepsis, a clinically relevant experimental model of this disease. Our findings suggest that at thermoneutrality (28℃ for rats), sepsis is associated with the development of fever, increase of plasma cytokine levels, and elevated hypothalamic PGE2 production and the mortality rate of animals was proportional to the severity of the disease. Conversely, in a cooler environment (Ta = 22 ℃), septic rats either maintained their Tb during mild sepsis or developed hypothermia during severe sepsis, with no observed mortality, despite the marked inflammatory response. We also demonstrate that the hability to regulate Tb, through mechanisms like tail skin vasoconstriction and brown adipose tissue thermogenesis, is influenced by both ambient temperature and the severity of sepsis. Specifically, HLI was reduced in all septic animals, regardless of Ta, while UCP-1 expression in brown adipose tissue increased only in mild sepsis under subthermoneutral conditions.
Despite the extensive research in experimental sepsis, there are several unsolved questions about the thermoregulatory dynamics during this disease. Body temperature was accessed in previous studies using the CLP method and both fever [21,22,23] and hypothermia [24,25,26] were observed, but none of these publications focused on the intrinsic body mechanisms associated with the changes in Tb and their relation with Ta.
Using the method of Romanovsky et al., which is based on the fact that rats have moderate tail skin vasodilatation in a neutral environment, we considered that Ta = 28 °C was thermoneutral and Ta = 22 °C was subthermoneutral in our experimental protocol. The thermoneutral zone (TMZ) consists of the range of Ta at which temperature regulation is achieved only by controlling heat loss and not by changes in metabolism [17]. At 28 °C, independently of sepsis severity, a fever response was observed, associated with a reduction of heat loss by the tail. It is also important to consider that the fever response might be influenced by the thermal gradient at this temperature, potentially making heat retention more effective. As BAT UCP1 expression was not altered in mild sepsis and had a decrease after severe sepsis, we infer that fever was mediated by tail vasoconstriction rather than an increase in non-shivering thermogenesis. Although we did not measure oxygen consumption as an index of non-shivering thermogenesis, a previous report corroborates our results, showing that CLP does not increase the expression of genes related to thermogenesis in the BAT, including Ucp1gene [26]. We speculate that cytokines such as IL-6, which has a thermogenic effect in chronic situations [27, 28], reduces BAT thermogenesis in acute conditions like sepsis due to its potent proinflammatory effects [29, 30]. These observations support the hypothesis that BAT thermogenesis is not the underlying mechanism for fever in our CLP model.
Similarly to what is observed in studies using different doses of LPS in a thermoneutral ambient [31], animals responded to sepsis with fever in a severity-dependent fashion. The degree of the increase in Tb can be also associated to the level of the inflammatory response and PGE2 synthesis. While circulating PGE2 produced by immune cells seems to be an initiator of fever, brain PGE2 is responsible for sustaining it [32]. In the hypothalamus, PGE2 produced by microglia and endothelial cells binds to EP3 receptors, an inhibitory receptor, expressed in glutamatergic neurons in the preoptic area which ultimately induces fever response [33, 34]. Supporting the febrigenic role of PGE2 in sepsis, it has been already shown that the administration of celecoxib, a selective COX-2 inhibitor, inhibited febrile response in septic rats after CLP [22]. Despite the importance of PGE2 in producing fever responses, fever observed during mild sepsis may be also result of a prostaglandin-independent pathway mediated by IL-1β [35]. A limitation of our study is the time point we analyzed the PGE2 levels, as PGE2 may be elevated earlier during mild sepsis but not after 12 h.
Fever is considered a defensive mechanism during an infection because it stimulates immune function and directly inhibits pathogens [36], but an exacerbated response can be deleterious to the organism. Dramatic increases in body temperature can lead to cell death mainly through the disruption of membrane stability and transmembrane transport, and the impairment of protein and DNA synthesis [37, 38]. In our experiments, an increased fever response was associated with higher mortality. Interestingly there was not an overlap of the peak of Tb (independently of the severity of sepsis) among the survivors (40.1 °C maximum) compared to non-survivors (40.5℃ or higher), suggesting that a point/range of Tb between these values is the highest temperature tolerated by septic rats. However, it is challenging to determine whether fever was the direct cause of death or a symptom of the harmful inflammatory response. It is likely that a combination of both factors contributed to organ dysfunction and subsequent death. The liver, for instance, is significantly impacted during sepsis, and markers of hepatic dysfunction are associated with mortality in the CLP model [39]. Previous studies have indicated that cytokines impair liver function during systemic inflammation [40, 41], but it has also been demonstrated that hyperthermia itself can be detrimental by reducing blood flow to the liver and causing hypoxia-induced hepatic damage [42]. Markers of heart [43, 44], kidney [45] and brain [46, 47] dysfunction are all increased during sepsis, even in the early stages of the disease, and are correlated to increased mortality.
When housed in a subthermoneutral environment (22℃), septic rats subjected to mild sepsis did not show changes in Tb, while those under severe sepsis had significant hypothermia. These findings are comparable to a study showing that the administration of the same dose of LPS provoked opposite changes in Tb when animals were in different ambient temperatures (fever in a thermoneutral Ta and hypothermia in a cool Ta) [31]. Thermoeffector recruitment was also distinct from a thermoneutral ambient. During mild sepsis, we observed a more consistent tail vasoconstriction and an increase in UCP1 expression in the BAT. The concomitant inhibition of heat loss and the increase of heat generation might have contributed to maintaining Tb at basal levels throughout the whole period of time analyzed, despite the substantially increased inflammatory response. On the other hand, in severe sepsis, UCP1 levels did not change and HLI was reduced only 10 h after CLP, which potentially contributed to the reduction in body temperature. These results point to a complex intercommunication between ambient temperature, infection severity, and thermoeffector recruitment. Interestingly, and aligned with our results, a study identified that lower ambient temperature was an independent predictor of hypothermia in septic patients [48].
Our data can be examined in light of an organism's dual protective strategies, i.e., resistance and tolerance, when facing infection, and how these strategies are influenced by Ta [49, 50]. Resistance involves an acute cellular immune response that actively attacks the pathogen, incurring significant metabolic costs. Tolerance, in contrast, suppresses the self-reactive immune response to limit tissue damage, thereby conserving energy for other physiological functions. A recent study [51] highlights this complexity showing that bacterial infection or LPS causes a trade-off with homeothermy and that infectious stress (at 22℃) induces hypothermia as an energy-conserving state. Our findings indicate that during severe sepsis, the host's response is distinctly modulated by Ta. At thermoneutral Ta, the organism favors a resistance strategy, leading to a dynamic but metabolically expensive immune response that increases mortality. Conversely, in a subthermoneutral Ta, the additional metabolic demand for BAT thermogenesis promotes a shift towards tolerance. This hypometabolic-hypothermic state is protective, reducing the deleterious effects of an overactive immune response. Interestingly, in mild sepsis, animals successfully controlled the infection regardless of Ta, evidenced by 100% survival. These results highlight the critical role of energetic trade-offs in determining the host's immune strategy, emphasizing the intricate crosstalk between homeothermy and immune function in managing energy consumption and optimizing survival outcomes during infection.
Previous studies have shown that metabolic stressors can significantly impact sepsis outcomes. For instance, Starr et al. [52] demonstrated that dietary restriction can improve survival rates in septic models by modulating Tb and the immune response, reducing inflammation. Other metabolic disorders like obesity [53, 54] and diabetes [55] also impact sepsis outcomes. The investigation of the impact of these circumstances during sepsis could provide valuable insights into the mechanisms of disease tolerance and resistance. Further research is needed to elucidate the underlying mechanisms of this crosstalk and its potential therapeutic implications for managing infectious diseases.
Despite the clinical significance of hypothermia during systemic inflammation, our understanding of its pathophysiology and the underlying mechanisms remains very limited compared to fever. PGE2, which has been extensively associated with fever, was also increased in the hypothalamus of hypothermic animals, probably as a consequence of the increased inflammatory response caused by CLP. Four subtypes of PGE2 (EP) receptors have been identified and while the activation of EP1 or EP3 receptors increases body temperature, administration of a EP4 receptor agonist decreases Tb [56]. The dynamics of the interplay between different prostaglandin receptors during sepsis and how it can lead to fever or hypothermia remains to be deciphered. We speculate that other mediators could also play a role in sepsis-induced hypothermia. Previous studies have shown evidence that prostaglandin D2 (PGD2), for example, is involved in hypothermic responses after LPS injection [57, 58].
We reinforce that we do not aim to suggest a duality between good vs. bad body or ambient temperatures during sepsis. Despite recent advances in the investigation of Tb changes and the outcome of sepsis, there is still no consensus if fever or hypothermia would result in a better prognostic in patients [59, 60]. Previous studies have reported different results from ours, with lower mortality rates after CLP in animals housed at thermoneutral Ta [61] and indicating that lower Ta exacerbates neuroinflammation [62] and cardiac dysfunction [63] in models of endotoxemia. Our data, however, point out that hypothermia can be protective during severe sepsis. Although it is often perceived as a thermoregulatory failure of the organism during an infection, it has been shown that Tb reduction in septic patients is a transient and non-terminal response [59]. Additionally, experimental studies have shown that this is a regulated process, i.e., a physiologically controlled phenomenon just like fever [64, 65]. These contradictory results highlight the complexity of this field of investigation and may be explained by differences in experimental methods. Variations can include the model of systemic inflammation used (such as CLP or LPS), the severity of the immune challenge (e.g., dose of LPS, number/ size of cecal punctures), the timepoint of analysis (early versus late phases of infection), the animal species (rats or mice), among other factors (sex, age, etc.). Translationally, these conflicting results seem to suggest that stratification of septic patients (by age, disease severity or metabolic status, for example) could be a better approach to investigate if cooling or warming would be a beneficial therapeutic effect.
Our investigation has some limitations. We only analyzed thermoregulatory alterations during the early stage of sepsis, specifically within 12 h after CLP. Although a previous study indicated that septic animals return their body temperature to basal levels 24 h even after severe CLP [22], it would be valuable to investigate how thermoeffectors are recruited and how Ta impacts survival in the later phases of infection. Additionally, we did not evaluate the disease progression across different severities and ambient temperatures over time. Implementing a scoring system to access the evolution of sepsis in our experiments could provide more comprehensive data to associate with mortality outcomes.
Finally, we would like to highlight the findings by Helbing et al. [66] where the authors reviewed recent publications (2019–2022) regarding sepsis research and observed a notable disregard of ambient temperature, which is often not even specified in publications. Our results revealed that changes in Ta may lead to completely different physiological responses and alter mortality during sepsis, and thus is relevant variable to be considered in experimental studies. In order to have comparable results and consistent findings we highly recommend that investigators always control and specify the ambient temperature in their protocols and publications when using experimental models of systemic inflammation.
Perspectives and conclusion
Our results indicate that the thermoregulatory mechanisms activated during sepsis are highly dependent on the severity of the condition and the surrounding environment. For instance, the protective role of hypothermia observed in severe sepsis within a subthermoneutral environment suggests a potential adaptive mechanism that could be therapeutically harnessed. This finding supports the growing interest in using hypothermia as a therapeutic strategy in the clinical practice [67,68,69]. Moreover, our data suggest that thermoregulatory responses, particularly the dynamics of fever and hypothermia, could serve as important biomarkers for assessing sepsis severity and prognosis. This could pave the way for developing more refined diagnostic tools that incorporate body temperature patterns as part of the criteria for stratifying sepsis patients, potentially leading to more personalized treatment approaches. Additionally, we emphasize the importance of considering ambient temperature in modulating immune response and survival outcomes during sepsis, underscoring the necessity of accounting for environmental factors in clinical settings.
Our results also demonstrate the necessity of accurately controlling ambient temperature in preclinical investigations. The current version of the Guide for the Care and Use of Laboratory Animals by the National Research Council in the USA, for example, recommends a housing temperature of 20–26 °C for mice and rats, a range that is below their thermoneutral zone. Prolonged exposure to a cold environment not only increases energy expenditure, but also leads to an increase in norepinephrine (NE) release, which acts on BAT to induce thermogenesis. However, NE's actions on the organism are not confined to BAT and have systemic effects, including on immune cells [70]. Chronic stress is associated with worse outcomes in autoimmune diseases and low-grade inflammation, for example [71, 72]. These changes in immune function should be carefully considered during experimental sepsis research. The impact of the length of exposure to a subthermoneutral temperature prior to infection has not been explored, but understanding this could provide important information regarding the necessary acclimation period for animals.
In conclusion, our experimental results have significant implications for the conceptual understanding of sepsis pathophysiology and translational relevance for diagnosis and therapy. By embedding our findings within the broader context of sepsis research, we demonstrate that both environmental and metabolic factors play critical roles in the pathophysiology of sepsis. This integrated approach not only advances our knowledge of the disease mechanisms, but also paves the way for developing innovative diagnostic and therapeutic strategies that could benefit a broader audience, including both clinical practitioners and researchers.



Comments (0)