
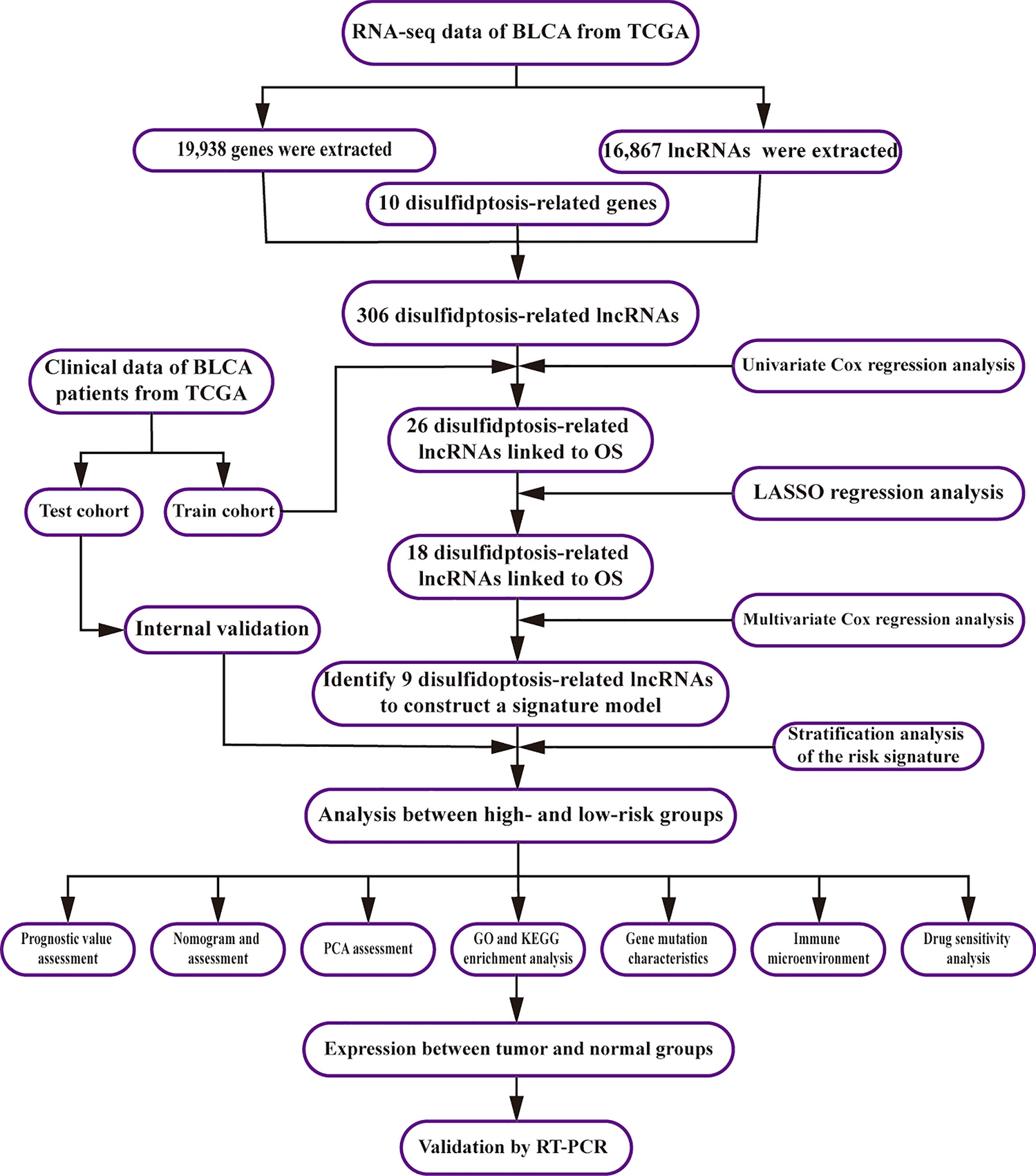
Remember me

SNU182 and Huh7 cells were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 units/mL penicillin, and 100 mg/mL streptomycin (Gibco) in a humidified incubator at 37 °C containing 5% CO2. To construct SR cells, SNU182 and Huh7 cells were first exposed to sorafenib at 5% of IC50 concentration. Then, the concentration of sorafenib was gradually increased at 10% of IC50 until the maximum tolerated doses was reached. To maintain the acquired resistance, SR cells (SNU182-SR and Huh7-SR) were cultured continuously in DMEM containing 1 μM concentration of sorafenib.
2.2 Construction of stable cell lines with overexpression or downregulation of DDX11-AS1Recombinant lentiviruses containing full-length DDX11-AS1 or DDX11-AS1 shRNA or the control were constructed according to the standard protocol. Cells were administered with 2 × 106 transducing units of lentiviruses and then subjected to selection with 2 μg/ml puromycin for a duration of two weeks. The target sequence of DDX11-AS1 shRNA was used as a previous study [20].
2.3 Cell transfectionThe transfections were carried out utilizing the Lipofectamine 2000 package (Invitrogen) in accordance with the instructions provided by the manufacturer. 48 h after transfection, cells were used for further investigation.
2.4 Cell viability detectionCell Counting Kit-8 (CCK-8; Dojindo) was used to assess cell viability according to the manufacturer’s instructions. 5 × 103 cells were seeded in 96-well plates with three replicate per group and treated with the corresponding concentrations of sorafenib for 24 h. Then, 10 μl CCK-8 regent was added into each well and incubated for 1.5 h. The absorbance at 450 nm was assayed by microplate reader (Thermo Fisher Scientific). The half-maximal inhibitory concentration (IC50) was acquired by nonlinear regression in GraphPad Prism 9.0 software.
2.5 Colony formation assay2 × 103 cells were plated in each well of 6-well plates, with three replicates per group. The cells were cultured in DMEM supplemented with the specified drugs for a period of two weeks, with the drug-containing medium being refreshed every 72 h. After the culture period, the colonies were fixed with 4% paraformaldehyde and stained with 0.004% crystal violet. The clones were then photographed and counted.
2.6 Measurement of cellular iron, malondialdehyde (MDA) and glutathione (GSH)The evaluation of lipid oxidation was evaluated by measuring the concentration of MDA in cellular lysates through the utilization of Lipid Peroxidation MDA Assay Kit (S0131, Beyotime) in accordance with the guidelines provided by the manufacturer. The levels of GSH were determined using the GSH and GSSG Assay Kit (Beyotime, S0053) following the protocol provided by the manufacturer. The intracellular Fe2 + concentration was quantified utilizing the Cell Ferrous Iron Colorimetric Assay Kit (Elabscience, Wuhan, China) in accordance with the manufacturer's guidelines.
2.7 Detection of ROSThe evaluation of lipid oxidation was evaluated using Reactive Oxygen Species (ROS) Fluorometric Assay Kit (E-BC-K138-F, Elabscience) in accordance with the manufacturer’s instructions. Then, values of ROS were measured using a fluorescence microplate reader (excitation wavelength 485–515 nm, emission wavelength 510–550 nm).
2.8 Western blotThe proteins were separated using SDS-PAGE and then transferred to a PVDF membrane. The membranes were blocked by 10% BSA and incubated with primary antibodies at a dilution of 1:1,000 overnight. Then, the membranes were incubated with Horseradish peroxidase-conjugated antibodies to rabbit IgG or mouse IgG (each at a dilution of 1:10,000, Jackson ImmunoResearch Laboratories). The protein bands were detected using the SuperSignal West Pico Kit (Thermo Fisher) following the manufacturer's instructions. The original images of western blot were shown in Supplemental Fig. 1.
Fig. 1
DDX11-AS1 is increased in the sorafenib-resistant HCC cells A. The IC50 values were determined for both parental and sorafenib-resistant (SR) Huh7 and SNU182 cells, which were exposed to sorafenib in a gradient concentration manner for 24 h. B. The expression of DDX11-AS1 in both parental and sorafenib-resistant (SR) Huh7 and SNU182 cells was tested using qRT-PCR assay. n = 3 per group, *p < 0.05
2.9 Isolation of nucleus and cytoplasmThe isolation of cellular nucleus and cytoplasm was performed using NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher) in accordance with the manufacturer’s instructions.
2.10 RNA isolation and quantitative real-time PCR (qRT-PCR)The Trizol reagent (Invitrogen) was utilized to isolate the entire RNA. The M-MLV Reverse Transcriptase (Invitrogen) was employed to generate first-strand cDNA. qRT-PCR was conducted in the StepOne™ Real-Time PCR System (Applied Biosystems, Foster City, USA) with SYBR® Green (Takara, Dalian, China) and the gene-specific primers. The target RNA level was quantified based on the 2−ΔΔCT value normalized the ACTB mRNA. Primer sequences for the genes are listed in Supplemental Table 1.
2.11 RNA immunoprecipitation (RIP) assayThe EZ-Magna RIP RNA Binding Protein Immunoprecipitation Kit (17–701, Millipore, USA) was utilized to conduct the RIP assay. Cells were grown until they reached 80–90% confluence, and then harvested by scraping. For each RIP reaction, 100 μl of cell lysate from approximately 2.0 × 107 cells was required. Subsequently, 5 μg of purified antibodies or corresponding IgG were added to the 100-μl cell lysate, and the mixture was rotated overnight at 4 °C. The RIP assay employed anti-Nrf2 (ab62352, Abcam), anti-Keap1 (10,503-2-AP, Proteintech) and normal rabbit IgG (PP64B, Millipore). The immunoprecipitated RNA was purified and analyzed with qRT-PCR.
2.12 co-immunoprecipitation (co-IP) assayco-IP was performed by lysing cells with an IP lysis buffer (P0013, Beyotime) and incubating up to 5 mg of total protein with 50 ml of Protein G-agarose suspension (Millipore, 16–266) for 3 h at 4 ℃ on a rocking platform to minimize non-specific binding. The primary antibodies were added to the supernatant and incubated for an additional 3 h at 4 ℃. Each immunoprecipitation mixture was then supplemented with 100 ml of Protein G-agarose and incubated overnight at 4 ℃ on a rocking platform. The immunoprecipitates were collected by centrifugation and washed three times with cold TBS. The agarose was boiled after adding the loading buffer and subjected to western blot assay. To prevent denaturation of heavy and light chains from antibodies used in immunoprecipitation assays, EasyBlot anti-mouse (GTX221667-01) or EasyBlot anti-rabbit (GTX221666-01) IgG HRP-conjugated secondary antibodies (Genetex) were utilized.
2.13 Statistical analysisAll assays were replicated independently with comparable outcomes at minimum three times. Statistical assessment and graphical representation of data were executed utilizing GraphPad Prism 9.0 software. The data were presented as mean ± standard deviation (SD). Unless otherwise specified, distinctions between two groups were evaluated by Student’s t-test, and contrasts between multiple groups were assessed by one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. p < 0.05 was deemed statistically significant.
Comments (0)