
Remember me
CAUTION: As this protocol involves handling and manipulation of radioactive material and or animals, researchers should consult with their home institution’s Radiation Safety and /or Medical Physics Department and undergo the required training prior to initiating this work. Steps should be taken to minimize exposure to ionizing radiations, including wearing whole-body and ring dosimeters. Gloves should be worn at all times. Radioactive waste MUST be disposed according to institutional radiation safety and local waste management guidelines. Existing home institution SOPs (Standard Operating Procedures) MUST be strictly followed for your own safety and that of your coworkers.
Cyclotrons, generator systems, linear accelerators (LINACs) or nuclear reactors can be used as a source of radioactive isotopes for this purpose. In either of these cases, access to a radiochemistry facility is essential. Radioisotopes and radioactive probes suitable to label immune cells need to be produced with high radiochemical purity (RCP; general consensus is ≥95%, however tracer dependent) and yield (RCY; tracer dependent). To achieve adequate radio-incorporation of the radionuclide into the targeting vector molecules, several radionuclide precursor solution considerations must be taken into account, including the radionuclide specific activity as the activity per unit mass of a radionuclide (radioactivity/mg) [21], possible trace contamination with elemental impurities that may compete for radio-incorporation with the radionuclide of interest, and the chemical form of both radionuclide itself and its formulation. In most instances, this information can be obtained by reviewing the Certificate of Analysis (CoA) that has been provided by the radionuclide manufacturer along with the material. Also, because the chemical form of the radionuclide solution may change over time, as a general rule, the time between radiochemical isolation of the radionuclide by the radionuclide producer and radiolabeling should be minimized. For more detailed radioisotope considerations and radiochemical probe design, refer to Volpe et al. [22].
Cells isolation protocols and culture medium may vary depending on immune cell type (T lymphocytes [23], NK (natural killer) cells [24], Tregs [25, 26] (regulatory T cells), gamma delta T cells [27], and dendritic cells [28]. Some cell types can be isolated using more than one protocol (see Hoerster et al. and Sato et al.) [24, 29]. For further details on different isolation protocols, practical isolation steps, required reagents, materials, and equipment, we refer the reader to commercially available kits.
Prior to labeling, cells are in vitro expanded, counted, and washed with phosphate buffer saline (PBS; Ca2+/Mg2- free) before being resuspended in PBS at room temperature (RT) (Note 1Footnote 1; Note 2Footnote 2)
1.Direct (ex vivo) Labeling
1.1.Add to 1-10x106 cells the desired radioisotope of choice (Note 3Footnote 3; Note 4Footnote 4; Note 5Footnote 5). To obtain optimal labeling, preliminary optimization and calculation of incorporation rates are required.
1.2.Incubate resuspended cells for 10-30 min at 37 degrees Celsius and 5% CO2 on agitating rack (at 350 rpm) (Note 6Footnote 6). Incubation time may vary depending on cell type and the probe.
1.3.Wash cells three times in ice-cold PBS (not containing Ca2+/Mg2-). Centrifuge and collect each wash solution in three separate collection tubes labeled “supernatant”, (wash 1” and “wash 2”, respectively.
1.4.Cells are resuspended in growth medium or PBS for further in vitro and/or in vivo experiments.
1.5.Radioactivity in resuspended cells and respective washes is measured using a gamma-counter measuring the radioisotope of choice (Note 7Footnote 7).
1.6.Radioactive cell-labeling efficiency is calculated using Equation 1.
$$LE\ \left(\%\right)=\left(\frac\right)x100$$
(1)
1.7.To evaluate the long-term radiotracer retention, radiolabeled cells are cultured at a confluence of 0.5x106 cells/mL medium. Samples are centrifuged at 1500 rpm and supernatants are removed and collected in prelabeled tubes. Cell pellets are re-suspended in their culture medium. Cell-associated radiotracer and supernatants are measured at the gamma counter and percentages are calculated using Equation 1. As cells continue to divide, expect a label/signal dilution over-time as the label will redistribute to the daughter cells. The percentage of total activity bound to the cells will decrease with time compared to the initial measurement according to radiotracer decay (the latter depending on half-life of the chose radiotracer), but cell expansion will not be detectable as the number of individually labeled cells will decrease [3], making the latter method unsuitable for a reliable long-term observation of fast-dividing cells.
IMPORTANT: Any commercially available gamma counter will provide decay corrected counts. In the unlikely circumstance that is not the case, a sample of known activity from the initial radioactive stock can be re-measured alongside with experimental samples. In lieu of these options, radioactivity at any given time can be calculated using the standard formula:
Or its derivatization:
where A is the activity, A0 is the initial activity, At is the activity after any time t, and λ is the decay constant, which for most radioactive material is readily available through the literature, or can be otherwise calculated using the following formula:
IMPORTANT: Not all radioactive probes are well suited candidates for the direct labeling of cell-based immunotherapies. One example is provided by Jacob et al., reporting on dose-dependent changes in polyclonally expanded human Tregs phenotype (downmodulation of CD4 and CD25), impaired proliferation both in vitro and in vivo, and failure to survive when labeled with 89Zr-oxine [31]. Always perform downstream assays to assess the consequences of direct radiolabeling, including viability, phenotyping, proliferation, functional and DNA damage studies. Also consider using a different PET radiotracer with a similar half-life (days) but with a potentially less harmful radioactive decay than 89Zr for this specific immune cell type.
2.Indirect (reporter gene-based) Labeling Method:
2.1.Viral Particles Generation
2.1.1.Viral constructs (Note 8Footnote 8) are generated bearing the following components: (i) a “regulatory complex” (Note 9Footnote 9); (ii) a “reporter gene”; (iii) a targeting receptor (e.g., CAR); (iv) a selection marker (e.g., either a fluorescent protein or an antibiotic resistance)
2.1.2.Either highly efficient viral-producing cells (i.e., 293RD [39, 40], and Phoenix Ampho) [41] or stable cells clonally selected for higher titer (i.e., PG13) [42] can be purchased through the American Type Culture Collection (ATCC) (Note 10Footnote 10)
2.1.3.The day prior transfection, seed cells in either 6-well plate or 10 cm plate to achieve 50-70% confluence on the next day (gently shake the plate to avoid uneven distribution) (Note 11Footnote 11)
2.1.4.On the next day, proceed with preparing the transfection mix by adding the required plasmid DNA and the transfection reagent (e.g., X-tremeGENE™, Lipofectamine™, Polyethyleneimine (PEI)). Always maintain sterile conditions
2.1.5.Incubate the transfection mix for 10 min at RT
2.1.6.Replace the growth medium of the virus producing cells with fresh one (without L-glutamine or antibiotics)
2.1.7.Gently add the transfection mix to the cells without disturbing them
2.1.8.Incubate plates at 37 °C and 5% CO2. Then, inspect the plates using a tissue culture microscope and gently replace the media with fresh medium (without L-glutamine or antibiotics)
2.1.9.48h later, the viral supernatant is ready to be harvested (please follow step 2.2.3 onwards)
2.2.Cellular immunotherapies transduction
This is a general protocol for immune cells transduction. Immune cells are non-adherent cells and therefore transduction protocols will require the use of non-tissue culture treated plates and pre-coating with RetroNectin. Transduction is performed on a 6-well format with 1 x 106 cells per well but both plate format and number of cells can be scaled up or down if needed.
2.2.1 Pre-coat a culture plate with RetroNectin (add at least 1mL per well in a 6-well plate to ensure complete surface coverage; scale quantities up or down depending on the chosen plate format)
2.2.2 Preserve plate sterility and prevent RetroNectin evaporation by sealing it with parafilm. Then store overnight at 4 °C or for 2h at RT
2.2.3 Harvest viral containing supernatant from virus producing cells and filter them through a 0.45 μm syringe filter (Note 12Footnote 12)
2.2.4 Replace media in the virus producing cells for subsequent transduction rounds
2.2.5 Supplement viral containing media with interleukin-2 (IL-2). Final concentrations used may vary depending on immune cell type (e.g., for T cells, 20 IU/mL; for Tregs 1000 IU/mL; for gamma delta T cells 300-1000 IU/mL) (Note 13Footnote 13)
2.2.6 Remove RetroNectin coating by aspiration and wash wells with 1 mL PBS supplemented with 10% FCS and leave at RT for 30 min
2.2.7 Collect immune cells (Note 14Footnote 14) and proceed counting them with using either an hematocytometer or an automated cell counter
2.2.8 Take the desired cell number for transduction and centrifuge at 500 x g for 5 min at 4 °C
2.2.9 Resuspend pellet in filtered viral containing supernatant at 1 x 106 cells/3 mL and incubate for 15 min at RT (Note 15Footnote 15)
2.2.10 Remove PBS (without Ca2+/Mg2+) with 10% FCS by aspiration and wash once with regular PBS (without Ca2+/Mg2+)
2.2.11 Aliquot 3 mL of supernatant containing 1 x 106 cells per well.
2.2.12 Perform spinoculation by centrifuging the plates at 100 x g for 1 h at 4 °C (Note 16Footnote 16)
2.2.13 Incubate plates at 37 °C and 5% CO2
2.2.14 Repeat transduction after 4-6 h. Repeat again on the next day for a total of four transductions (Note 17Footnote 17)
2.2.15 Before repeating the transduction, centrifuge the plate at 500 x g for 5 min
(a)Repeat steps 2.2.3 to 2.2.5
(b)Remove about 2 mL of old viral supernatant by aspiration and add 3 mL fresh filtered one
(c)Add fresh filtered viral supernatant to the cells and continue from step 2.2.12
2.2.16 48h after the last transduction round, spin plates at 500 x g for 5 min
2.2.17 Gently aspirate supernatant containing viral particles
2.2.18 Wash cells twice in PBS (without Ca2+/Mg2+)
2.2.19 Resuspend cells in their fresh culture medium supplemented with IL-15 (Note 13), move into a larger flask and keep in the incubator at 37 °C and 5% CO2
To ensure high viral titer and subsequent efficient transduction of immune cells, we recommend optimizing the Multiplicity of Infection (MOI; representing the number of viral particles required to achieve a 100% transduction). To calculate the MOI, use the following Equation 5:.
2.3.In vitro characterization of transduced cellular immunotherapies:
2.3.1.In vitro assessment of successful transduction can be done by flow cytometric analysis if the reporter bearing construct also includes a fluorescence marker (e.g., if reporter is co-expressed with Green Fluorescent Protein (GFP), it can be detected by flow cytometry). For accurate quantification of reporter expression, we recommend performing quantitative flow cytometry (QFCM). If expression is too low (<95%), select positive clones by fluorescence-activated cell sorting (FACS). If the reporter-based vector also includes an antibiotic resistant gene, the corresponding antibiotic is used as a selection marker in the cultured media to maintain the population pure (>99% reporter+). Remember to use an appropriate negative control (parental non-transduced cells) to determine flow cytometer settings. If/when available, we suggest to also use a positive control (e.g., a similar cell type previously transduced with the same transgene, thus offering expression of the same FP (fluorescent protein)
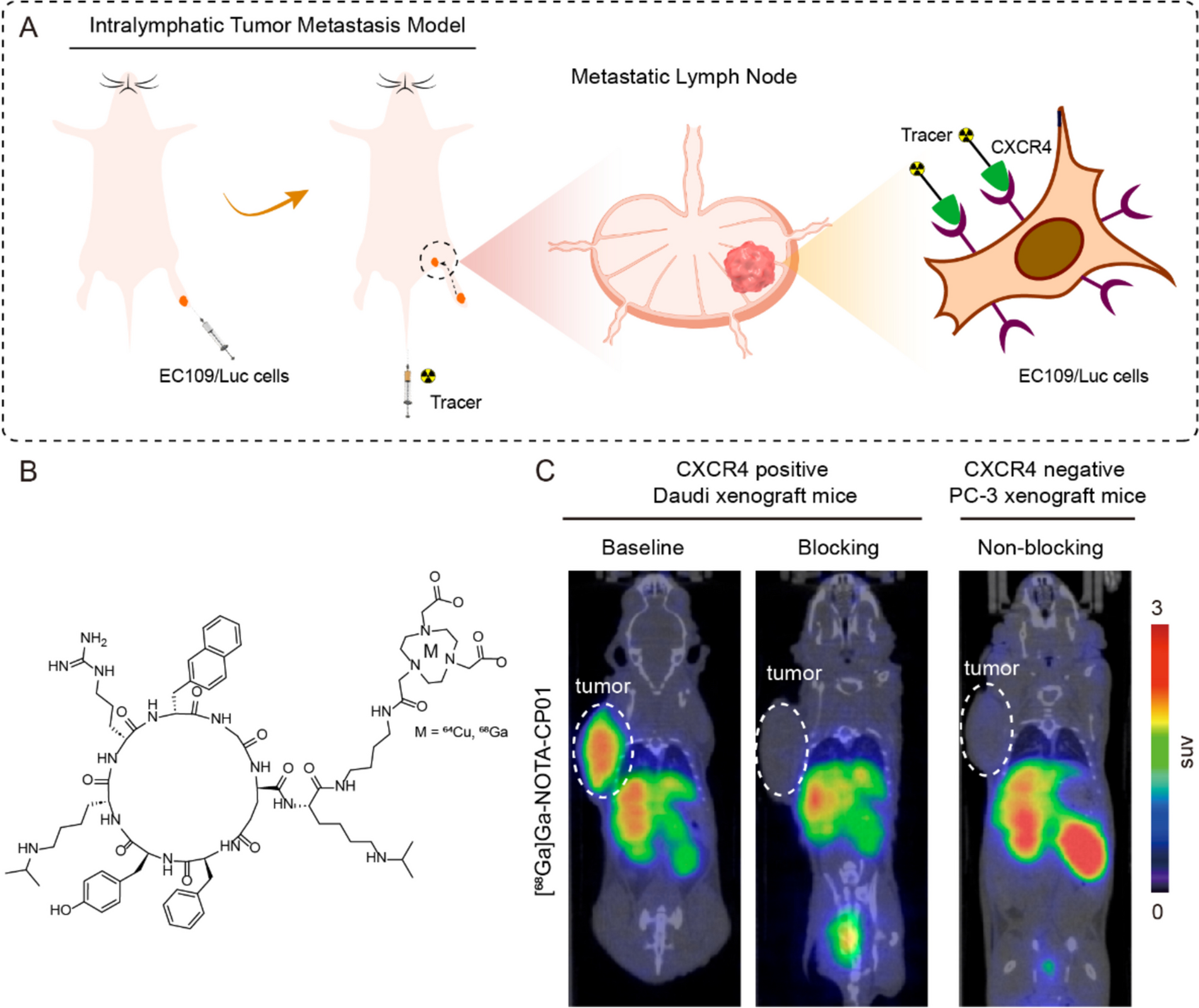
2.3.2.In vitro assessment of reporter function implies performing a radioactive uptake assay. The type of radioisotope varies depending on the chosen reporter gene [5]
2.3.2.1.Aliquot 1 x 106 cells in pre-labeled Eppendorf tubes called “cells”. Prepare samples in triplicates and include also “control samples” corresponding to non-transduced parental cells (Note 18Footnote 18)
2.3.2.2.Wash cells twice with ice-cold PBS
2.3.2.3.Incubate cells with desired amount of radioactivity (suggested is 1.35-1.50 μCi per sample) in 1 mL PBS for 30 min-1 h at 37 °C and 5% CO2 (Note 2)
2.3.2.4.Blocking or competitive studies as “specificity controls” can be performed depending on the reporter (e.g., sodium perchlorate is used as a competitive substrate for the sodium iodide symporter NIS and resulted in uptake depletion demonstrating radioactive uptake specificity) [48]. In that case, pre-treat cells with blocking or competitive agent, then add it again with the radioactivity (keep concentration constant throughout the assay). We encourage to also add a negative control (parental non-transduced cells) to determine basal uptake levels
2.3.2.5.Centrifuge samples at 1500 rpm at RT, collect 100 μL of supernatant and transfer it into a pre-labeled tube named “supernatant”. Safely remove the remaining 900 μL and dispose of it according to institutional radiation safety guidelines
2.3.2.6.Wash the cells twice with ice-cold PBS alternating centrifugation steps. Collect 100 μL from each washing solution and transfer them into pre-labeled tubes named “wash 1” and “wash 2”, respectively
2.3.2.7.Follow steps 1.4 to 1.5.
2.3.2.8.Calculate radiotracer uptake using the following Equation 6, where CPM represents the decay-corrected radioactivity counts per minute (Note 19Footnote 19)
$$\% Uptake=\left(\frac\right)\ x\ 100$$
(6)
New emerging cell therapeutics (including human cardiac progenitor cells and human-induced pluripotent stem cells (iPSCs)) are also amenable to indirect labeling, and step-by-step protocols describing their engineering using the sodium iodide symporter NIS reporter gene are available [49, 50].
IMPORTANT: When the chosen reporter gene is a transporter or a surface protein, localization studies must be performed (confocal microscopy) prior to the radioactive uptake assay (as described in 2.3.2) to prove its correct localization as a fundamental pre-requisite for its correct function (imaging capability shown by the successful uptake of desired radioactive probe). Staining controls (e.g., membrane marker) and negative controls (parental non-transduced cells) are required (see Volpe et al. as an example) [47].
IMPORTANT: Whether Direct or Indirect Labeling was performed, we strongly recommend the reader to perform all application-dependent downstream experiments, including assessing the effects of cell labeling on their long-term viability, proliferation, phenotype, functional status (including activation markers and/or CAR expression), cytotoxic capability and potential radiation- induced DNA damage as compared to the parental cells (see referenced examples) [24, 31, 47]. These are crucial steps to determine whether the labeling procedure had detrimental effects on the cellular function and are a sole responsibility of each user. Perform these experiments alongside a negative control (parental non-transduced cells). Other controls may be needed depending on the type of experiment and its complexity and will therefore not be discussed in this protocol.
3.In vivo tracking of cell-based immunotherapies in relevant preclinical models:
IMPORTANT: Prior to animal testing, ensure that the in vivo protocol is approved by the local Ethical Review Panel and is compliant with the institutional ethical and safety guidelines and regulations. Laboratory personnel is required to undergo training before performing invasive procedures in living animals. Access to an imaging facility is essential to perform the following protocol.
Relevant therapeutic models will have to be established prior to starting the labeling of therapeutic cells. The choice of genetic backgrounds and animal strains (i.e., humanized, fully immunocompetent or with various levels of immune compromisation) will depend on study goals and is a sole responsibility of the researcher (see Duncan et al. [51] for CAR-T cell models; for a general overview of available murine models for cancer immunotherapy, refer to Olson et al. [52]).
3.1.Ex vivo radiolabeled cells:
3.1.1.Further to the washing (as described in step 1.3), centrifuge radiolabeled cells at 1500 rpm for 5 min at RT
3.1.2.Remove radioactive supernatant without disturbing the cell pellet and safely dispose of it
3.1.3.Resuspend cells in 50-200 μL saline or serum-free medium (Note 20Footnote 20)
3.1.4.Keep cells on ice until in vivo intravenous administration
3.1.5.Aliquot a sample of radiolabeled cells and washes for further in vitro testing (i.e., gamma counting and radiotracer retention; see steps 1.5 and 1.7 of 1.1, respectively)
3.1.6.Prepare animals for injection. If interested in the distribution kinetics of directly labeled therapeutic cells once in the body, a dynamic scan will be required. Therefore, animals must be anesthetized beforehand (please follow steps described in Note 25) in an induction chamber with 1.5-2.0% (v/v) in O2 (flow rate 1-1.5 L/min) (Note 21Footnote 21)
3.1.7.Assess anesthetic depth using either the tail pinch or the pedal reflex method
3.1.8.Place the mouse onto a heating pad to maintain physiological temperature and with its nose in an anesthetic supply mask and proceed with warming the tail using an infrared light lamp
3.1.9.Slowly inject radiolabeled cells intravenously via tail vein catheter or using a syringe connected to a hypodermic needle (gauge 29-31) (Note 20 and Note 22Footnote 22)
3.1.10.If no dynamic imaging is needed post injection, perform intravenous injection of radiolabeled cells into conscious animals (skip steps 3.1.6 to 3.1.8). However, to this date, no published literature is available describing changes in the biodistribution of any cell-based therapeutic product upon administration under anesthesia.
3.1.11.If imaging is needed post injection, place the mouse onto the bed of a microSPECT-CT or microPET-CT scanner and ensure that anesthetic supply is constant, and animal is asleep
3.1.12.Ensure correct positioning of the animal (e.g., sphinx-like position)
3.1.13.Before starting the imaging, ensure that animal monitoring devices are installed (including an ECG pad to record the electrocardiogram and a rectal temperature probe for temperature monitoring)
3.2.Reporter gene labeled cells:
3.2.1.Start with animal preparation (see steps 3.1.6 to 3.1.8 of “Ex vivo radiolabeled cells” subsection)
3.2.2.Intravenously inject therapeutic cells expressing the desired radionuclide-based reporter into anesthetized mice (Note 22)
3.2.3.If no imaging is needed post injection, perform intravenous injection of radiolabeled cells into conscious animals (skip steps 3.1.6 to 3.1.8)
3.2.4.If imaging is needed post injection, inject the reporter-paired radioactive probe, and allow blood clearance prior to imaging (Note 23Footnote 23)
3.2.5.Follow steps 3.1.11 to 3.1.13
3.2.6.To observe the early stages of cell immunotherapies distribution, inject the radioactive probe immediately after the cells. If prolonged monitoring is needed, the radioactive probe can be administered at the desired time point and repeated imaging can be performed indefinitely (Note 24Footnote 24)
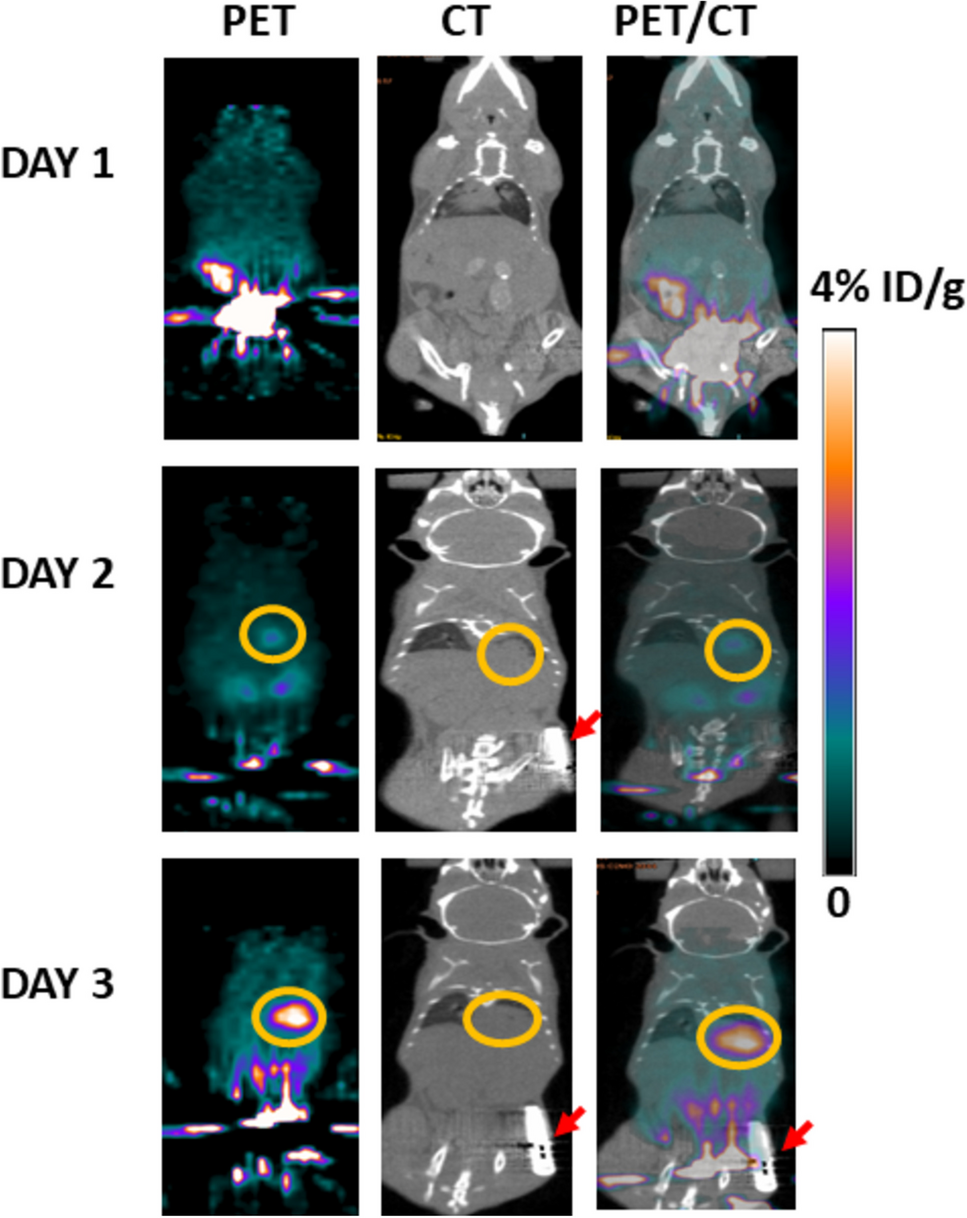
4.Nuclear imaging by microSPECT-CT or microPET-CT and data analysis:
Parameters such as in vivo distribution, metabolism and clearance from the body may vary depending on the radioactive probe used (refer to Volpe et al. for preferential distribution and properties of available radioactive probes) [22]. Therefore, it is important to add some waiting time prior to imaging in order to allow radiotracer in vivo biodistribution and avoid high blood signal.
Depending on the radiotracer characteristics (gamma rays or positron emitter), either SPECT (Single-photon emission computed tomography) or PET (Positron emission tomography) will be used.
4.1.Set the desired CT parameters (e.g., 55 kVp tube voltage, 1200 ms exposure time with one-degree angular stepping, 360-degree projections). When ready, acquire CT image to provide anatomic information and accurate tissue attenuation correction of SPECT or PET
4.2.If performing SPECT imaging, set the parameters for image acquisition (e.g., for 99mTc[TcO4-], perform a 45 min scan, 40 frames, 360-degree projections, collimator pinhole size 1 mm, 110-150 keVp energy window). Then acquire SPECT image
4.3.If performing PET imaging, set the parameters accordingly (e.g., for [18F]BF4-: perform a 30 min scan, 1:5 coincidence mode, 400-600 keVp energy window). Then acquire PET image (Note 25Footnote 25 and Note 26Footnote 26)
4.4.If repeated animal imaging is needed, allow animals to fully recover from anesthesia and transfer them into a maintenance unit
4.5.If this is a terminal imaging procedure, euthanize animals according to approved protocol (see Shomer et al. [55])
4.6.Reconstruct the SPECT- or PET-CT images using a 3D fully iterative Monte Carlo-based algorithm
4.7.Ensure that images are calibrated to the injected radioactivity and corrected for tissue attenuation, dead time and radioisotope decay (Note 27Footnote 27)
4.8.Save the data in the standard exchange format for medical images “DICOM” (Digital Imaging Communication in Medicine) and load them in the selected image analysis software (Note 28Footnote 28)
4.9.Before proceeding with image-based analysis, ensure that the CT and SPECT or PET are correctly co-registered
4.10.Delineate regions of interest (ROIs) and quantify corresponding radioactive signal (Note 29Footnote 29 and Note 30Footnote 30)
4.11.Non normalized results can be expressed as percent injected dose (%ID). Normalized results can be expressed as (i) percent injected dose per volume (%ID/mL), when considering the individual organs/tissues volumes, or (ii) standardized uptake value (SUV), with the latter accounting for the average radioactivity across the whole animal (Note 31Footnote 31).
4.12.Express image data as either %ID (Equation 7) or %ID/mL (Equation 8). To best convey the quantitative information and to greatly facilitate the comparison of images across different studies, it is important to apply the same intensity scale (Note 32Footnote 32). The corresponding intensity scale bar should be always present for each figure. For more guidelines on radionuclide-based image analysis, refer to Weber et al. [58] For those with little to no experience in nuclear-based image analysis, refer to a recently published protocol from Cawthorne et al. [59]
5.Ex vivo biodistribution:
5.1.Following to step 4.5, measure the whole animal radioactivity and note the value and time of measurement
5.2.Proceed with animal dissection and harvest relevant organs and tissues
5.3.Measure the radioactivity in the tail and in the remaining carcass post dissection (remember to take note of values and time of measurement) (Note 31)
5.4.Selected organs and tissues are collected in pre-weighted tubes and weighted
5.5.Prepare radioactive calibration standards with a known activity to simplify decay correction:
5.5.1.On the day of the experiment, prepare standards from the same stock solution used for radiotracer injection
5.5.2.Prepare duplicate samples in 1.5 mL Eppendorf tubes
5.5.3.Each duplicate will be read at the gamma counter before and after your experimental samples using the same settings (i.e., exposure time and energy window)
5.5.4.At the end of the measurements, a calibration curve (activity vs counts per second) can be generated by linear regression
5.6.Measure radioactivity in all selected organs and tissues alongside with radioactive calibration standards using an automated gamma counter (note the time of measurement) (Note 33Footnote 33)
5.7.Express values as %ID (Equation 7), %ID/g (Equation 8 Equation 8) or SUV (Equation 9), where the latter are calculated using the following formulas:
$$\% ID=\left(\frac\right)x\ 100$$
(7)
$$\% ID/g=\left(\frac\right)x\ 100$$
(8)
$$SUV=\left(\frac\right)$$
(9)
Comments (0)