
Remember me
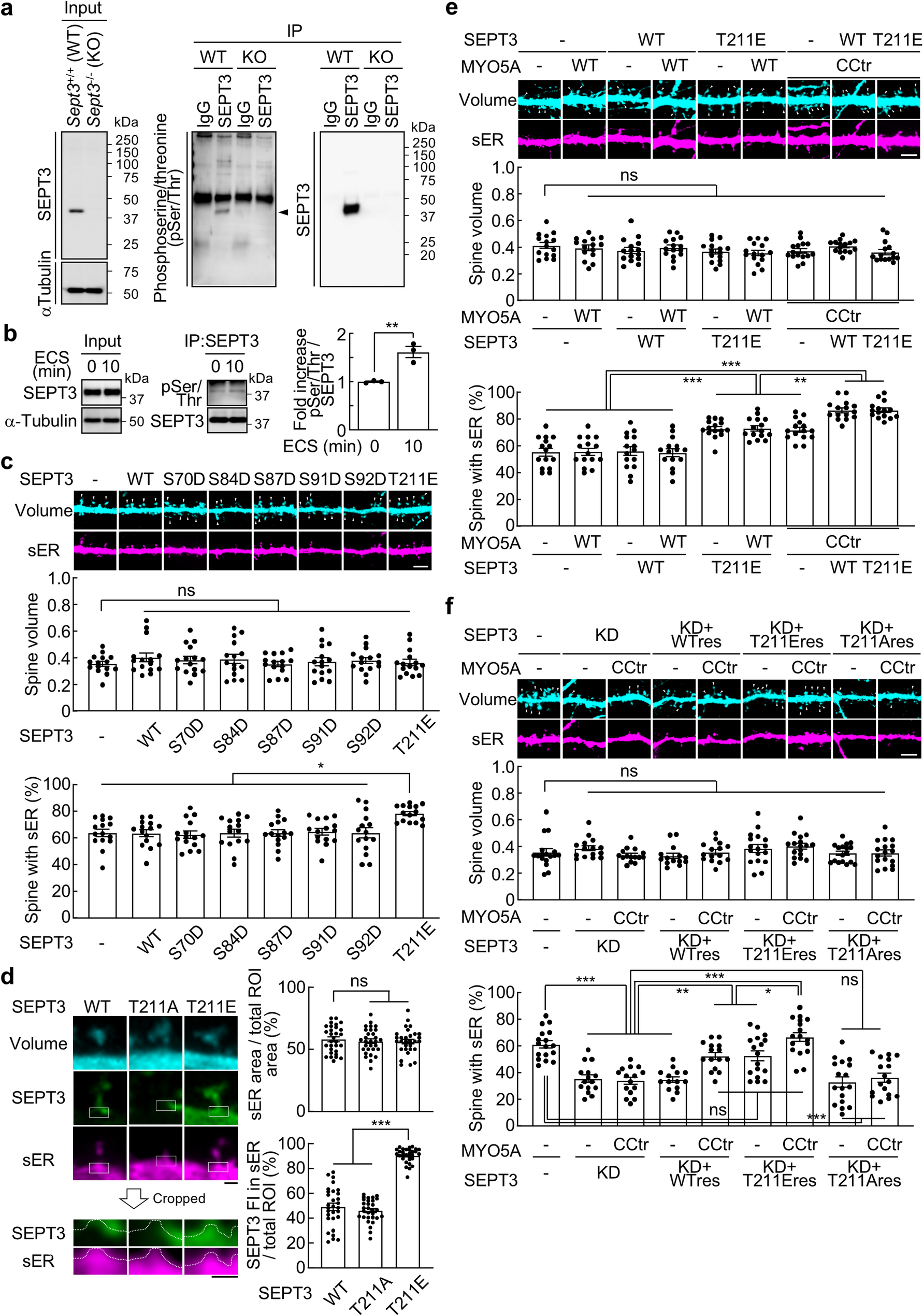



We sequenced cDNA synthesized from total mRNA from the cerebellum, cortex, hippocampus, and striatum of 20-week-old male and female (n = 5 each) C57BL/6J mice using an ONT GridION device (Fig. 2A). We obtained 85,909,493 reads passing quality control metrics (Methods), with each brain region receiving at least 16 million reads across the ten samples for that region (Fig. 2C). The hippocampus had the lowest number of total reads (n = 16,739,487), potentially due to our reduced starting material as it is smaller than the other brain regions we assayed. We aligned and quantified our data using the nf-core [16] nanoseq pipeline and Bambu [17], a tool for performing machine-learning-based transcript discovery and quantification of long-read RNA-sequencing data with high precision and recall [18]. When visualizing our samples based on variance-stabilization transformed (VST) gene counts by principal component analysis (PCA), samples are separated by tissue (Fig. 2B). The difference in cerebellum samples to all other brain region samples drove the greatest gene expression variation in the data set (PC1, 33% of the total variance, Fig. 2B).
Fig. 2
Long-read Nanopore RNA sequencing across four mouse brain regions. (A) Overview of the study design. (B) PCA plot (PCs 1 and 2) of VST gene counts. Here, we colored samples by brain region. (C) Bar graph of the total number of long reads sequenced for each tissue. (D and E) Bar graphs of the number of novel and annotated genes and transcripts. (F) Histogram of the transcript counts per gene, truncated to 25 transcripts per gene. Supplementary File 2 includes the numbers of all transcripts measured for each gene
Next, we determined any potentially novel genes and transcripts we had captured with Bambu [17]. We identified 285 genes and 382 transcripts not previously annotated in mouse GENCODE release M31 (Fig. 2D and E) and considered them “novel.” These 382 potentially novel transcripts correspond to 354 unique genes. Of the 382 novel transcripts, 309 (81%) transcripts belonged to novel genes, and 73 (19%) belonged to previously annotated genes (Additional File 1). Interestingly, when we examined the overall expression distributions of these novel transcripts compared to annotated transcripts, novel transcripts were expressed significantly more than annotated transcripts (one-sided Wilcoxon rank sum test, p = 7.850072e-114, Additional File 8 - Supplementary Fig. 1A), potentially due to the stringent expression thresholds Bambu has to identify novel transcripts. Of these novel transcripts, 279 out of 382 (79%) had a mean counts per million (CPM) of at least one across all samples. To test if transcript discovery thresholds contributed to the effect that novel transcripts were expressed higher, we performed Wilcoxon rank sum tests at four mean CPM thresholds (1 CPM, 2 CPM, 5 CPM, and 8 CPM - top 10%, Additional File 8 - Supplementary Fig. 1B-E), and the result was still significant each time (One-sided Wilcoxon rank sum tests, p < 1e-11). Therefore, on average, novel transcripts were still expressed more than annotated transcripts. This differs from other studies that identified novel transcripts using long read data, though these studies used different transcript discovery tools [19,20,21], and bambu consistently discovers fewer false positives [18]. However, all genes had a mean of 3.2 transcripts, while novel genes had a mean of 1.1 transcripts, though a subset of all genes (n = 76) had over 25 transcripts expressed (Fig. 2F, Additional File 2). Two long non-coding RNA (lncRNA) genes, Gas5 and Pvt1, had the most transcripts (149 and 129, respectively). In short, we generated a lrRNA-seq dataset for four brain regions and both sexes of C57BL/6J mice, in which we identified potentially novel genes, transcripts, and patterns of gene expression variance across mouse brain regions.
Differential gene expression and differential transcript expression and usage identified across brain regionsWe calculated DGE and DTE using the R package DESeq2 [22], a ‘gold-standard’ method that performs consistently well for DTE in long-read sequencing data [18]. We show a cartoon example representing significant DGE between two conditions in Fig. 3A. We found 8,055 (Wald test with Benjamini-Hochberg (BH) correction p < 0.05) pairwise brain region DGE events involving 3,546 unique genes (Fig. 3B), where the cerebellum, compared to the striatum, had the most DGE (n = 2,229, Wald test with BH correction p < 0.05), and the cortex, compared to the hippocampus, had the least DGE (n = 349, Wald test with BH correction p < 0.05) (Fig. 3B). Consistent with our PCA (Fig. 2B), each brain region compared to the cerebellum had the most DGE, with 920 genes consistently differentially expressed in the cerebellum compared to the other regions (Fig. 3B). We calculated DTE for each expressed transcript, and we considered a gene to have DTE if it had at least one transcript with differential expression for that comparison (a cartoon example representing significant DTE between two conditions is in Fig. 3C). We identified 11,138 DTE events (Wald test with BH correction p < 0.05) associated with 4,126 unique DTE genes (Fig. 3D). Unlike DGE, the greatest difference in DTE genes was between the cerebellum and cortex (n = 2,620, Wald test with BH correction p < 0.05), and the least was between the cortex and the hippocampus (n = 345, Wald test with BH correction p < 0.05) (Fig. 3D).
Fig. 3
DGE, DTE, and DTU across pairwise brain region comparisons. Cartoon representations of a gene with three isoforms (actual genes may have more or fewer isoforms) exemplifying (A) differential gene expression (DGE - violet), (C) differential transcript expression (DTE - turquoise), (E) and differential transcript usage (DTU - green). UpSet plots of the overlap of genes with (B) DGE (Wald test with BH correction p < 0.05), (D) DTE (Wald test with BH correction p < 0.05), and (F) DTU (t-test with BH correction p < 0.05) between pairwise brain region comparisons. The bar plot above denotes intersection size, circles denote which comparisons have overlap, and the set size reflects the total number of genes with DTU for that comparison. We omitted intersections of fewer than 40 genes from the chart for legibility for panels B and D. We omitted intersections of fewer than five for legibility in panel F. (G) Stacked bar chart representing pairwise brain region comparison overlap across DGE, DTE, and DTU. Genes included in the chart must express at least two transcripts
Next, we calculated DTU for each pair of brain regions using the DTU method SatuRn [23] with the R package IsoformSwitchAnalyzeR [24]. We show a cartoon example representing significant DTU between two conditions in Fig. 3E. Here, we considered a gene to be a DTU gene if it had a t-test statistic (calculated from the log-odds ratio and variance of the quasi-binomial generalized linear model) BH-corrected p-value < 0.05 for at least one of its transcripts where genes had at least two expressed transcripts (Methods). We analyzed DTU across brain regions and found 1,051 DTU events in 648 unique genes (Fig. 3F). The most DTU genes were in the cerebellum compared to the striatum (n = 355, t-test with BH correction p < 0.05), and the least were in the cortex compared to the hippocampus (n = 31, t-test with BH correction p < 0.05) (Fig. 3F). Consistent with our other analyses (65% for DGE and 71% for DTE), we identified the majority of DTU genes (66%) from comparisons including the cerebellum (Fig. 3B, D, F). Interestingly, the number of DTU genes (n = 63, t-test with BH correction p < 0.05) shared across all three comparisons including the cerebellum was a smaller percentage (10%) of the total unique DTU genes (Fig. 3F) than DGE (26%) or DTE (25%) (Fig. 3B, D), suggesting that DTU analysis is less driven by the cerebellum. We also directly compared which genes were identified for each analysis (DGE, DTE, and DTU) that expressed at least 2 transcripts and qualified for DTU analysis. We found that DGE and DTE genes had the most overlap across comparisons, with a small proportion of significant genes for each comparison identified by all three methods (Fig. 3G).
We also performed functional enrichment analysis using gprofiler2 [25] of DGE, DTE, and DTU genes for all comparisons (Additional Files 3–5). For the cortex compared to the cerebellum DGE, DTE, and DTU genes, we found enrichment (Fisher’s exact test with g: SCS correction, p < 0.05) for 1742, 2431, and 54 terms. Strikingly, we found a much larger percentage of terms associated with the neuronal synapse in DTU (24/54, 44%; e.g., synapse, glutamatergic synapse, post-synapse, synaptic signaling, neuron-to-neuron synapse, and postsynaptic membrane) compared to DGE (50/1742, 2.9%) and DTE (77/2431, 3.2%). Because a larger proportion of DTU genes were enriched for pathways required for synaptic neurotransmission, this suggests that DTU potentially identifies biologically distinct molecular signatures from DGE and DTE. To see if any ontology terms were enriched in genes specific to DTU, we performed functional enrichment analysis on the DTU genes that did not have significant DGE or DTE. We identified multiple ontologies enriched for genes with DTU (Fisher’s exact test with g: SCS correction, p < 0.05), including histone deacetylation, TCF/WNT signaling, cytoplasmic ribosomal proteins, Kir4.1-dystrophin complex, and muscle-derived dystrobrevin-syntrophin complex (Additional File 6). The term with the most significant enrichment for DTU genes between cerebellum and cortex was histone deacetylation (adj. p = 0.014), suggesting that these genes (Mta1, Arid4b, and Suds3) play an integral role in isoform-specific chromatin remodeling between the two regions (Additional File 6). Overall, a pairwise comparison of DGE, DTE, and DTU between brain regions revealed marked heterogeneity for each analysis per comparison, with a greater overlap in DGE and DTE than in either analysis with DTU. This underscores that isoform usage may be masked when only considering differential expression, hiding biologically distinct molecular signatures.
DTU sex differences are brain region-specificDue to known sex biases in healthy brain gene expression [2] and in brain-related disease phenotypes [8, 9], we asked if there were sex-biased DGE, DTE, or DTU events by brain region. First, we measured DTU across sexes, combining brain regions, and identified four genes with DTU: Zfp862-ps, Gm10605, Shisa5, and Zfp324 (t-test with BH correction p < 0.05) (Additional File 8 - Supplementary Fig. 2). Zfp862-ps and Zfp324 are a pseudogene and gene, respectively, for zinc finger proteins that contain a DNA-binding domain. While pseudogenes have traditionally been considered non-coding, they have been shown to regulate other genes and form viable proteins [26, 27]. Notably, the human ortholog of Shisa5, SHISA5, has been previously identified as having sex-biased splicing in human brain white matter [2], in line with our finding of sex-biased splicing in mouse brain regions. Finally, Gm10605 is a predicted lncRNA gene. We did not identify any of these genes in our within-brain region analyses, suggesting that for these genes, we were underpowered to identify DTU in each region alone.
We next calculated DGE, DTE, and DTU across sexes within each brain region (Table 1; Fig. 4). We identified 23 region-specific genes with DTU by sex (analysis of deviance chi-squared test with BH correction p < 0.05): 14 in the cortex, seven in the striatum, and two in the cerebellum (Table 1). Despite documentation of phenotypic sex differences in the hippocampus [28], we did not find sex DTU in the hippocampus (Fig. 4C). None of the 23 genes overlapped between brain regions, suggesting these sex differences may be brain region-specific. When we compared these DTU genes to DGE genes for each region, none overlapped (Fig. 4A-D), and only three of 23 overlapped with DTE. Therefore, by analyzing DTU, we identified 20 additional genes with differential sex effects.
Table 1 Genes with brain-region-specific DTU across sexesFig. 4
DGE, DTE, and DTU across sex within brain regions. (A-D) Euler diagrams represent the overlap of genes with significant DGE (Wald test with BH correction p < 0.05, purple), DTE (Wald test with BH correction p < 0.05, cyan), and DTU (analysis of deviance chi-squared test with BH correction p < 0.05, green). The brain regions represented are (A) cerebellum, (B) cortex, (C) hippocampus, and (D) striatum. (E) Switchplot displaying a transcript summary, gene expression, isoform expression, and isoform usage of the gene Anxa7 across female (F; light color) and male (M; dark color) cerebral cortex. In the indicated comparison, ns denotes not significant, * denotes P < 0.05, ** denotes P < 0.01, and *** denotes P < 0.001
We highlighted one of these sex-significant cortex DTU genes, Anxa7, for its many known connections to sex-associated phenotypes in humans (Fig. 4E). Human ANXA7 is a member of the annexin family, and humans express this gene in all tissues [29]. ANXA7 has multiple links to sex hormones; for example, ANXA7 promoter activity is affected by estrogen and progesterone nuclear receptors [30]. In addition, patients with schizophrenia express this gene lower than healthy controls [31]. In our study, we measured three distinct Anxa7 isoforms: ENMUST00000100844.6 (the Ensembl canonical transcript), ENMUST00000065504.7, and ENMUST00000224975.2 (Fig. 4E). When we aggregate transcript expression, Anxa7 does not have differential gene expression between males and females in any region. However, there was DTU of ENMUST00000065504.7 and ENMUST00000100844.6 across sex (Fig. 4E). Males expressed ENMUST00000100844.6 (the only transcript that included exon 5) higher than females. Humans have a documented clinical variant of uncertain significance (gnomAD variant 10-75143086-T-A) in the conserved male-biased exon [32]. Strikingly, 11/16 reported cases with this variant were in XY males and only 5/16 in XX females [32]. In the alternatively spliced exon 5, multiple transcription factor binding sites exist, including for FOXO1, which is strongly sex-associated and a key transcription factor associated with early pregnancy [33]. In summary, analysis of sex-significant DTU genes revealed differential isoform usage by sex within brain regions that would have otherwise been undetected by gene or transcript expression analyses, including genes with known sex-associated phenotypes.
There are two main patterns of sexually dimorphic transcript usage: sex-divergent and sex-specificIn addition, we noticed distinct patterns in sex DTU genes expressing two transcripts (Fig. 5A-B). First, we identified sex-divergent switches, i.e., sexually dimorphic transcript expression, where a single dominant transcript switch is in the opposite direction for both sexes (Fig. 5A). We identified sex-divergent switches in Mtcl1, Sel1l, 6430548M08Rik, Srgn, and Lmtk3. For example, the sex-divergent gene Mtcl1 has two transcripts, ENMUST00000086693.12 and ENMUST00000097291.10, where ENMUST00000086693.12 is dominant in males and ENMUST00000097291.10 in females (Fig. 5C). Mtcl1 codes for Microtubule Crosslinking Factor 1 and is expressed highly in the cerebellum in the literature and our dataset [29]. Human MTCL1 is known to be essential for the development of Purkinje neurons [34]. Despite its connections to the cerebellum, we only saw DTU in Mtcl1 by sex in the cortex. We also identified sex-specific isoform switches, i.e., where one sex expresses one isoform, but the other sex had almost equal expression of both isoforms (Fig. 5B). We identified sex-specific isoform switches in Rab28, Fbxo25, Leprot, Kifap3, and Plppr2. Rab28 (Fig. 5D) has a female-specific isoform, ENMUST000000201422.4, which had approximately equal expression as the other isoform, ENMUST00000031011.12, in females, while ENMUST00000031011.12 was the only isoform expressed in males. RAB28 is an essential gene for vision, and loss of function mutations in RAB28 cause cone-rod dystrophy in humans [35, 36]. Thus, in addition to identifying significant differences in isoform usage between sexes, we also found distinct patterns of sex DTU gene expression, with sex-significant DTU genes showing either sex-divergent or sex-specific transcript expression.
Fig. 5
Sex-divergent and sex-specific DTU. (A-B) Representative cartoons exemplify two transcript expression patterns of isoform switching: sex-divergent (A) and sex-specific (B). (C) Switchplot displaying a transcript summary, DGE (Wald test with BH correction p < 0.05, purple), DTE (Wald test with BH correction p < 0.05, cyan), and DTU (analysis of deviance chi-squared test with BH correction p < 0.05, green) of the sex-divergent gene Mtcl1 in the cortex between females (F; light color) and males (M; dark color). (D) Switchplot displaying a transcript summary, DGE (Wald test with BH correction p < 0.05, purple), DTE (Wald test with BH correction p < 0.05, cyan), and DTU (analysis of deviance chi-squared test with BH correction p < 0.05, green) of the sex-specific gene Rab28 in the striatum between females (F; light color) and males (M; dark color). Please note that these plots do not display all possible transcript structures of this gene, only the ones measured in our dataset. In the indicated comparison, ns denotes not significant and * denotes P < 0.05
A web application for visualizing DGE, DTE, and DTU in mouse brain lrRNA-seq dataFinally, we built an R Shiny application for our data set. Users may create custom gene expression heatmaps (Fig. 6A) or examine switch plots for individual genes using the IsoformSwitchAnalyzeR package (Fig. 6B). We also provide the option for users to download the intermediate gene expression and isoform switch test result data and plots directly. Our Shiny application has been made publicly available at https://lasseignelab.shinyapps.io/mouse_brain_iso_div/.
Fig. 6
Shiny app presents a user-friendly interface for exploring our mouse brain dataset. Screenshots of our web application (A) The “Custom Gene Expression Heatmap” lets users examine and download the gene-level expression of any gene(s) of interest in our dataset. Users can also download the expression and isoform switch test result data to analyze further or download the plots as-is. (B) In the “Pairwise Brain Region Comparison” tab, users can visualize their gene of interest in pairwise brain region comparisons in real-time and download expression and isoform switch test result data and plots
Comments (0)