
Remember me






The clinicopathological information of 32 women diagnosed with IBC is summarized in Table 1. The age at diagnosis ranged from 29 to 82 years, with 14 patients aged 50 years or younger and 18 patients showing a family history of cancer. According to the Body Mass Index (BMI), 22 patients were overweight or obese (BMI ≥ 25). Twelve patients presented histological grade III, 19 had clinical stage III at diagnosis, and 13 were triple-negative (TNBC: ER/PR/HER2 negatives). Thirteen patients presented distant metastases at diagnosis and eleven during follow-up. Twenty-four women died at a medium time of 18.3 months (3.06 to 105.2 months), six were alive, and two died by other causes.
Patients at clinical stage III were initially treated with neoadjuvant chemotherapy with four cycles of doxorubicin and cyclophosphamide, followed by paclitaxel for 12 weeks. In HER2-positive patients, trastuzumab was administered, and some hormone receptor-positive IBC patients also received tamoxifen or anastrozole. Patients alive after the neoadjuvant treatment were referred to surgery and adjuvant radiotherapy. Five patients presented a pathological complete response (pCR) after the neoadjuvant treatment. Stage IV patients were treated with palliative therapy, including fluorouracil, doxorubicin, and cyclophosphamide.
Mutational profileThe mutational profile was performed in 28 IBC samples (21 previously reported) [9] using t-NGS (105 cancer driver genes). The most common variants were detected in TP53 (18 cases), BRCA2 (9 cases), and PIK3CA (6 cases) (Fig. 1E). Interestingly, 20 out of 28 cases showed variants in genes involved in the homologous recombination pathway (Table S2).
Fig. 1
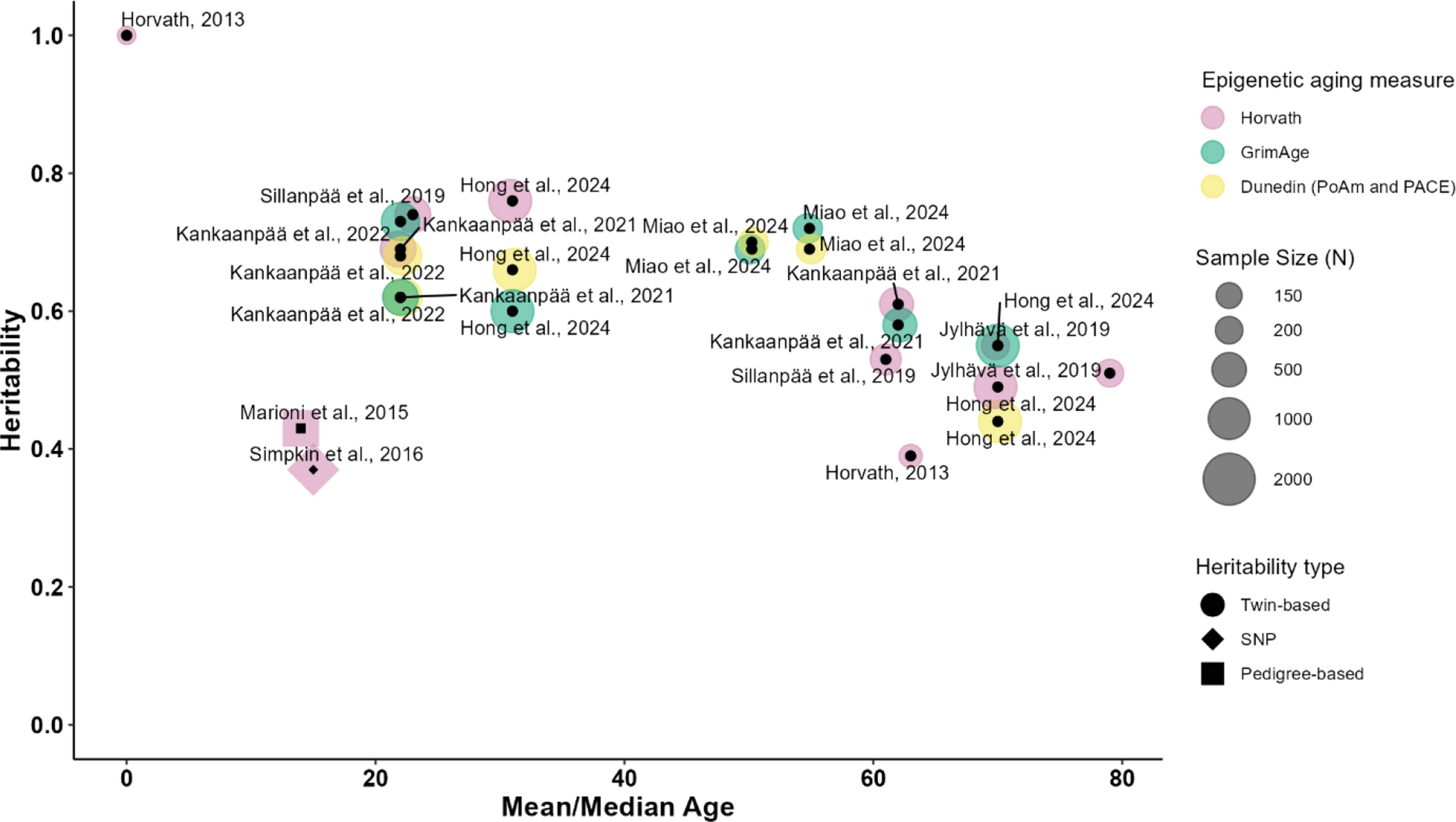
Methylation profile of the IBC internal dataset. A Distribution of the differentially methylated CpG probes between tumor and normal tissues (NT). The proportion of CpGs in relation to its gene location (TSS200/TSS1500, body/exons boundaries, intergenic regions, 5′UTR/first exon and 3′UTR); and to the CpG islands context (island, open sea, shelf, and shore) was based on the Illumina EPIC annotation. B Unsupervised K-means clustering analysis based on 46,908 identified DMPs revealed four clusters: cluster 1 is composed of adjacent normal samples and three clusters with IBC samples. Rows indicate the CpG sites, while columns represent samples. Clinical features of each case are represented below the heatmap along with targeted next-generation sequencing data for specific genes. The estrogen receptor (ER), progesterone receptor (PR), HER2 status, and the mutational pattern of TP53, BRCA1, BRCA2, and homologous recombination genes are indicated below the heatmap. C The survival curves (Kaplan–Meier and log-rank test) showed no significant statistical differences among clusters. D Univariate Cox regression analysis based on clinical variables and overall survival of 24 IBC patients evaluated by DNA methylation profiling. The forest plot shows the hazard ratios (squares), and the horizontal bars represent the range between the lower and upper limits of the 95% confidence intervals (CI) in the log2 scale. IBC patients positive for estrogen (ER), progesterone (PR), and or human epidermal growth factor receptor 2 (HER2) were taken as a reference against triple-negative tumors. In the analysis of the BMI variable, we excluded patients within the normal weight range due to the small sample size (N = 4); overweight patients were used as a reference. For the remaining variables, we considered the absence of the corresponding predictive factor as a reference for analysis (*p < 0.05). E A panel of 105 cancer-related genes was investigated in 28 IBC patients using t-NGS. Highlights include clinical, molecular, and vital status information. Genes are organized in descending order of alterations. The top bar plots illustrate the number of altered genes detected in each sample, and the percentages on the right indicate the number of samples with genetic alterations for a given gene among all analyzed samples. Genes without selected variants were excluded
Identification of DMPs and DMRs in IBCThe DNA methylation performed in 24 IBC and six adjacent normal breast samples (NT) revealed 46,908 DMPs (FDR < 0.05 and |∆β|≥ 0.2), in which 30,919 (66%) were hypomethylated and 15,989 (34%) hypermethylated (Fig. 1A). Hypermethylation was predominant in the CpG islands (39.9%), followed by open sea regions (32.7%), while most hypomethylation was found in the open sea region (80.8%). Hypermethylated sites (32.4%) were frequently found in CpGs mapped in gene bodies and hypomethylated DMPs in intergenic regions (42.1%). Hypermethylated CpGs mapped in promoter regions (21.2%) were more frequently observed than in hypomethylated DMPs (11.7%). Annotations related to shore, shelf, and other regions are illustrated in Fig. 1A. Supplementary Fig. 1 summarizes the study design and main findings.
The unsupervised clustering analysis of all DMPs revealed four clusters: One grouped all normal samples (cluster 1), and three had IBC samples (Fig. 1B, Table S3). Clinical and molecular features distribution, such as estrogen (ER) and progesterone receptors (PR), HER2 status, metastasis at diagnosis, TP53, BRCA1, BRCA2, and HR genes mutations were distributed among the three clusters (Fig. 1B). Cluster 3 was enriched with triple-negative cases, TP53 mutation, and cluster 4 with ER/PR positive, HER2-negative. Both clusters 3 and 4 were enriched with HR mutated genes. Although no significant difference was observed in the survival curves and BMI among these groups of patients, cluster 3 presented shorter survival (Fig. 1C; Table S4).
The Cox regression model was applied to evaluate the prognostic role of molecular and clinical information gathered from our cohort of IBC cases screened for DNA methylation. We found that TNBC (p = 0.036; HRatio = 1.131; CI 0.095–2.175), metastasis at diagnosis (p = 0.00048; HRatio = 2.23; CI 0.993–3.466), and HR mutated genes (p = 0.011; HRatio = 2.64; CI 0.588–4.700) were significantly correlated with overall survival as independent factors for increased risk (Fig. 1D). Multivariate regression analysis revealed that only metastasis at diagnosis (p = 0.0032; HRatio = 2.10; CI 0.704–3.487) remained a prognostic indicator for overall survival. A pairwise comparison of potential variables showed a robust association between triple-negative tumors (p = 0.015; HRatio = 1.45; CI 0.288–2.622) and metastasis at diagnosis (p = 0.0003; HRatio = 2.57; CI 1.178–3.969), indicating a poorer overall survival. Similar findings were observed in patients presenting metastasis at diagnosis (p = 0.0073; HRatio = 1.71; CI 0.460–2.953) and HR genes mutation (p = 0.0305; HRatio = 2.36; CI 0.222–4.499). Cox regression analysis highlighted the significant role of metastasis at diagnosis in predicting poorer overall survival. Furthermore, our model underscores that the separate interaction of this variable with TNBC and tumors with HR mutations is significantly associated with an increased risk of death.
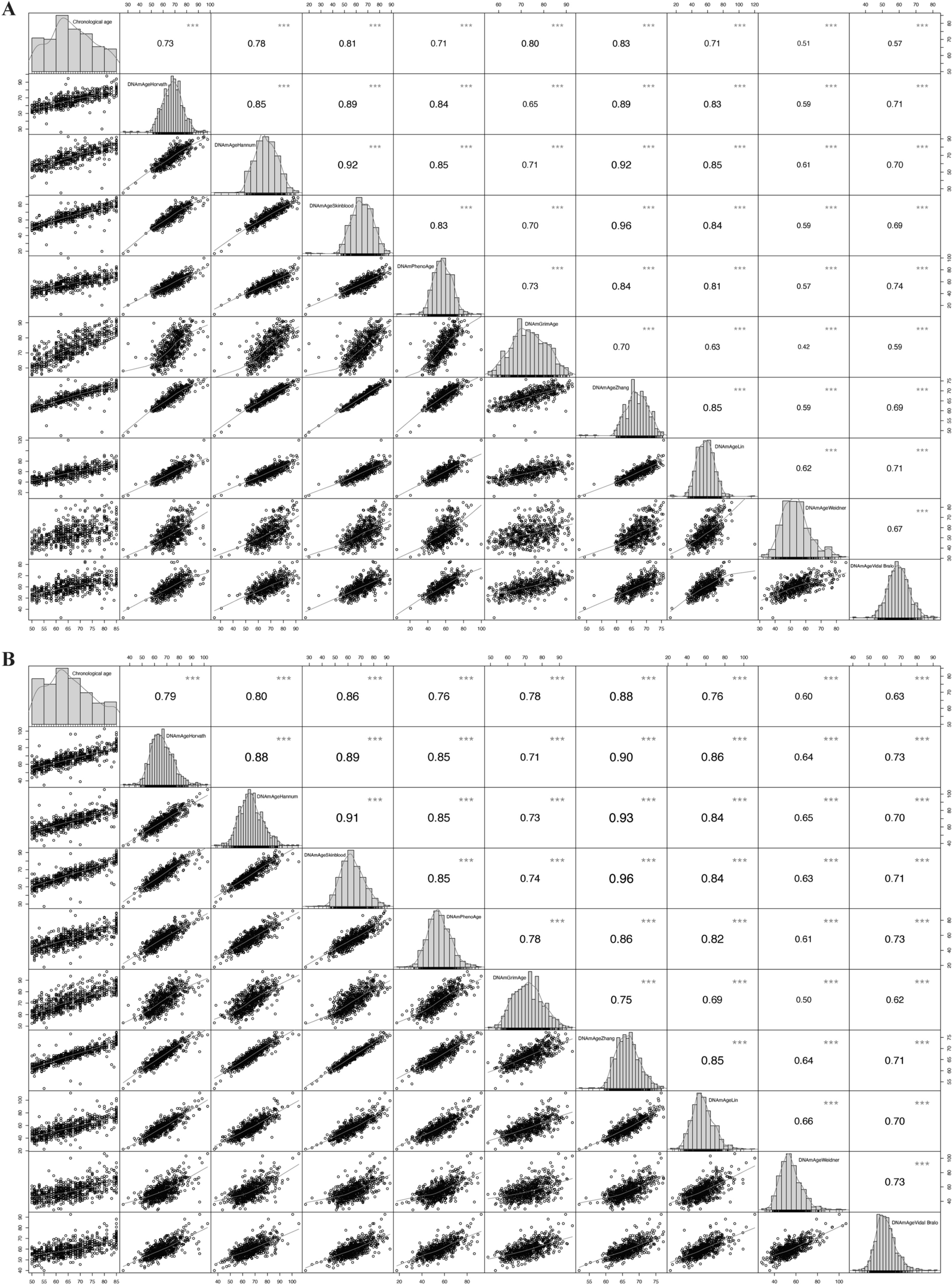
Among the 46,908 significant DMPs, 17,868 CpGs were mapped in intergenic regions and 29,040 (11,135 hypomethylated and 17,905 hypermethylated) in known genes. The DNA methylation data is available in the Gene Expression Omnibus database (GSE238092). The enrichment analyses showed pathways involved in signal transduction (MAPK signaling pathway, PI3K-Akt signaling pathway, Ras signaling pathway), cellular adhesion and movement (focal adhesion, ECM-receptor interaction, regulation of actin cytoskeleton, cell adhesion molecules), and neuronal system (axon guidance, neuroactive ligand-receptor interaction, glutamatergic synapse, synapse vesicle cycle, morphine addiction, and nicotine addiction), among others. The top 20 KEGG pathways are shown in Fig. 2A.
Fig. 2
Differentially methylated probes (DMPs) comparison performed among the internal dataset, 27 k Van der Auwera et al. (2010), and TCGA-BRCA advanced tumors. A KEGG pathway analysis shows five shared enriched pathways among three datasets. B Venn diagram obtained from dataset comparisons (internal dataset, TCGA-BRCA, and Van der Auwera et al., 2010) shows 385 shared DMPs among the datasets comparison
A total of 4369 DMRs mapped on known genes was found (2392 DMR hypomethylated and 1977 hypermethylated) (Table S4). Two genes presented more than 30 CpGs in the same DMR, TBX15 (36 CpGs) and OR2I1P (31 CpGs).
Comparison of DNA methylation profiles of IBC and non-IBC using external datasetsThe comparison of our findings with the Van der Auwera et al. (2010) [27] data (1353 DMPs; p < 0.00001, |∆β |≥ 0.17) revealed 417 shared DMPs with the same methylation status, 92 hypomethylated and 325 hypermethylated (Figure S1). Using the same criteria adopted in the internal dataset (FDR < 0.05 and |∆β|≥ 0.2) to analyze advanced breast tumors from TCGA, we found 38,662 DMP, in which 14,611 DMP were found in both datasets (6298 hypomethylated and 8313 hypermethylated). The comparison among these three datasets resulted in 385 shared DMPs (Fig. 2B) mapped in 333 genes, 69 hypomethylated and 264 hypermethylated (Table S5). Enrichment analysis revealed five main pathways: Focal Adhesion, Calcium Signaling Pathway, cAMP Signaling Pathway, Neuroactive Ligand-Receptor Interaction, and Protein Digestion and Absorption (Fig. 2A, Table S6).
The gene expression data of IBC samples retrieved from the GSE45581 (Whole Human Genome Microarray 4 × 44 K Agilent platform) was compared with our DNA methylation results (Pearson correlation, p < 0.05), revealing 151 DMPs mapped in 110 genes; 68 probes mapped in 50 genes presented a negative correlation (Table 2). The gene annotation using CancerMine (http://bionlp.bcgsc.ca/cancermine/) and MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/) databases showed 50 genes, of which 31 are cancer-related (Table 2). We also accessed the expression levels of these genes using the GSE207248. Ten genes were hypomethylated and overexpressed (such as the oncogenes CHST11, KIF26B, LAIR1, NTM, RUNX2, VOPP1, and WDR5), and six hypermethylated and down expressed (including the tumor suppressor genes CDO1, CTDSPL, EBF1, ROBO3, and TGFBR3) (Table 2).
Table 2 List of probes and genes with a negative correlation between the internal dataset and expression data from the GSE45584. Significant values of correlation are presented (p < 0.05)Locus-specific DNA methylation analysisBisulfite pyrosequencing was used to quantitatively determine the methylation of CGs mapped on BCAT1, CXCL12, and TBX15 genes. Figure 3 shows the mean DNA methylation levels of consecutive CpG dinucleotides flanking those interrogated by microarrays as well as the methylation levels of individualized DMPs. The analysis of gene body DMR encompassing 16 CpGs of BCAT1 gene confirmed increased methylation levels in IBC compared with normal samples. We also detected a trend to increase methylation levels in IBC versus normal tissues (p = 0.0725), but no differences were observed between triple-negative IBC compared with non-triple-negative IBC. The comparison with the expression levels of BCAT1 of TCGA-BRCA stages III-IV revealed no difference between non-TNBC and normal samples. However, a significant increase in expression was observed between normal versus TNBC and non-TNBC versus TNBC.
Fig. 3
Loci-specific DMRs analyzed by bisulfite pyrosequencing of CpGs associated with selected genes (BCAT1, CXCL12, and TBX15) using the internal cohort and cross-validation with gene expression levels using external cohorts (advanced III-IV stages from TCGA-BRCA and IBC from GSE45581). The first column shows the comparison of the means of DNA methylation levels of each associated DMRs containing the interrogated CpG probes and the flanking CpG dinucleotides between normal and IBC samples. Tumor samples were also dichotomized in non-TNBC and TNBC, and the methylation levels were compared. The DNA methylation levels of distinct DMPs confirmed higher methylation levels of BCAT1 and TBX15 genes and lower levels of CXCL12 in IBC. The methylation levels of TBX15 gene were associated with the subtype TNBC and obesity (*p ≤ 0.05, **p ≤ 0.01; ***p ≤ 0.001, and ****p ≤ 0.0001). ns: not significant (p > 0.05)
Based on the unsupervised K-means clustering analysis of DMPs and in the literature data, two differentially methylated CpGs (cg06702993 and cg11267527) of CXCL12 were evaluated. Triple-negative IBC showed a significant decrease in DNA methylation levels of CXCL12 compared to non-triple-negative samples (p = 0.0481). These findings are in accordance with the expression levels of CXCL12 of TCGA-BRCA stages III-IV, showing a decreased expression in advanced tumors compared to normal samples and TNBC.
We selected four differentially methylated CpGs extracted from the DMR of TBX15 (10 analyzed), which were also altered in TCGA-BRCA dataset. We confirmed increased methylation levels in IBC compared to normal samples. Triple-negative tumors presented increased methylation levels of TBX15 compared with non-triple-negative IBC cases. The expression level of TBX15 was confirmed as significantly decreased in advanced BC tumors (TCGA-BRCA stages III-IV) and IBC (GSE45581) compared to normal samples (Fig. 3G). In addition, we found a significant negative association between methylation levels of TBX15 and obesity (cg14565725, OR 0.9182, CI 0.8332 to 0.9864, p = 0.0004; cg07892597, OR 0.9391, CI 0.8661 to 0.9996, p = 0.0486; cg10703826 OR 0.9336, CI0.8690 to 0.9892, p = 0.0184; and cg24884142, OR 0.8864, CI 0.7909 to 0.9659, p = 0.0042).
Comments (0)