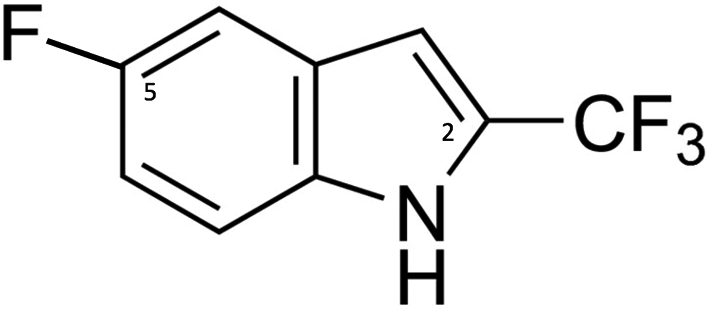
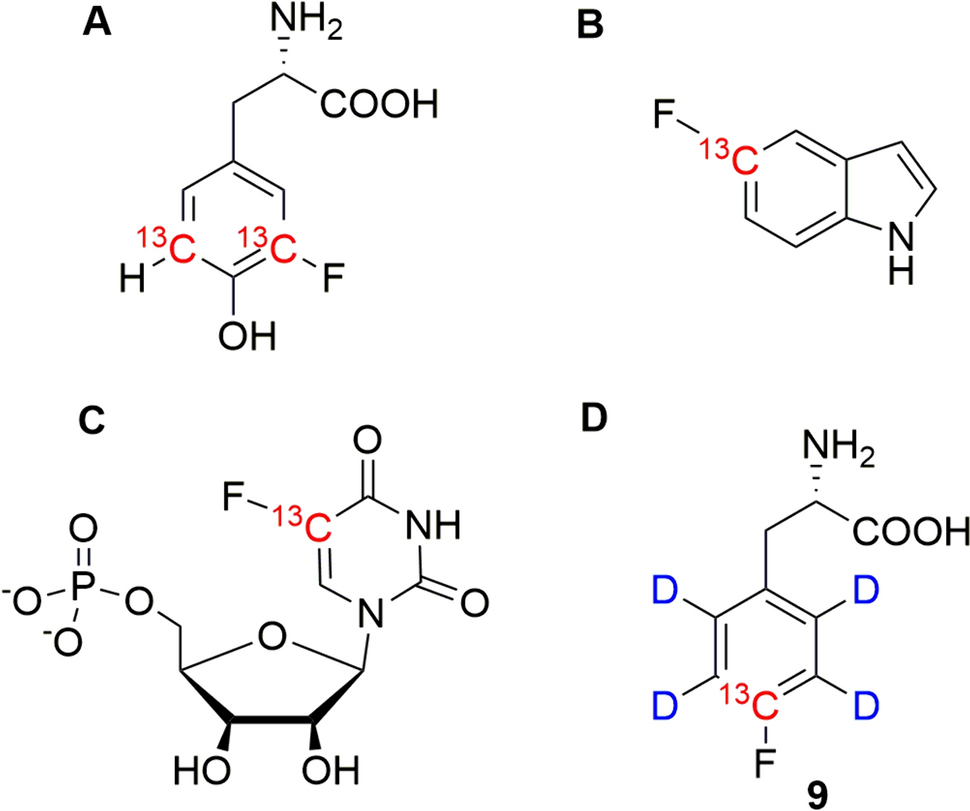
Synthesis of [4-13C, 2,3,5,6-2H4] 4-fluorophenylalanine 9
The synthetic route to access compound 9 is shown in Fig. 2A. [2-13C] acetone 1 with an isotopic purity of ~ 99% 13C was purchased from CortecNet® (France); N,N′-1,3-bis(2,6-diisopropylphenyl)chloro-imidazolium chloride/CsF (PhenoFluorMix®) was prepared according to literature (Fujimoto and Ritter 2015). Sodium nitromalonaldehyde was synthetized from mucobromic acid as described in literature (Fanta 1952). All other reagents were obtained from commercial suppliers and used without further purification. Microwave reactions were performed in an Initiator + from Biotage®. NMR-spectra of compound 9 and synthetic intermediates are shown in the supporting information (SI).
[1-13C] 4-nitrophenol 2
Compound 2 was prepared by condensation of commercially available [2-13C] acetone with nitromalonaldehyde (Lichtenecker 2014). An aqueous NaOH solution (4.4 g in 20 mL) was slowly added to a mixture of sodium nitromalonaldehyde (3.25 g) and [2-13C] acetone 1 (1 g) in H2O (200 mL) at 0 °C using a dropping funnel. After the addition was complete, the flask was tightly closed and stirred for 6 days at 4 °C. The resulting solution was cooled to 0 °C and 6 N HCl (26 mL) was slowly added. Filtration of the solution resulted in a dark solid, which was taken up in 6 N HCl (26 mL) and boiled gently for 10 min. The warm mixture was filtered, and the two combined filtrates were extracted with diethyl ether (6 × 100 mL). Subsequent drying of the combined organic phases over MgSO4 and evaporation of the diethyl ether under reduced pressure yielded a yellow solid. The crude product was purified over a silica gel chromatography column by elution with hexane–ethyl acetate (6:4 v/v). The reaction yielded 1.47 g (63%) of [1-13C] 4-nitrophenol 2. 1H NMR (400 MHz, CDCl3) δ: 8.19 (dd, J = 9.4 Hz, 2 H, CHarom.), 6.92 (dd, J = 9.4 Hz, J = 2.4 Hz, 2 H, CHarom.), 5.39 (1H, OH); 13C NMR (100.6 MHz, CDCl3) δ: 160.94 (13CH), 126.16, 115.63 (d, J = 68.0 Hz).
[1-13C, 2,6-2H2] 4-nitrophenol 3
A microwave vessel was loaded with [1-13C] 4-nitrophenol 2 (338 mg), D2O (1.5 mL), and DCl 7 N (0.5 mL). The mixture was irradiated for 1 h at 170 °C. The product was then extracted with diethyl ether (3 × 60 mL). The organic phases were dried over MgSO4, and the solvent was evaporated, resulting in 330 mg (96%) of [1-13C, 2,6-2H2]4-nitrophenol 3, isolated as yellow crystals. 1H-NMR spectroscopy analysis indicated quantitative deuterium incorporation at positions 2 and 6. 1H NMR (400 MHz, CDCl3) δ: 6.92 (d, J = 9.6 Hz). 13C NMR (100.6 MHz, CDCl3) δ: 160.94 (13CH).
[4-13C, 3,5-2H2]4-fluoronitrobenzene 4
1.52 g of CsF and 0.68 g of N,N′-1,3-Bis(2,6-diisopropylphenyl)-2-chloro imidazolium chloride were stirred at 140 °C in vacuo for 3 h. The resulting solid mixture was allowed to cool to room temperature (RT) and merged with 142 mg of [1-13C, 2,6-2H2]4-nitrophenol 3 in a round bottomed flask equipped with a reflux condenser under argon atmosphere. Dry toluene (7 mL) was added, and the mixture stirred at 110 °C for 24 h. After completion, the reaction mixture was cooled to RT and filtered through celite, eluting with dichloromethane. The filtrate was then concentrated in vacuo to yield a red oil, which was further purified using a chromatography column with heptane/ethyl acetate (20:1) as the eluent. This step resulted in the isolation of 110 mg (76%) of [4-13C, 3,5-2H2]4-fluoronitrobenzene 4. 1H NMR (400 MHz, CDCl3) δ: 8.29 (dd, JHF = 4.5 Hz, JCH = 10.8 Hz).13C NMR (100.6 MHz, CDCl3) δ: 166.4 (d, J = 257.8 Hz).
[4-13C, 3,5-2H2]4-fluoroaniline 5
114 mg (1 mmol) of substrate 4 were dissolved in 4 mL of anhydrous methanol in a Schlenk flask and 40 mg of 10% Pd/C catalyst were added. The flask was purged with argon and then exposed to H2 using a hydrogen balloon. The mixture was stirred under H2 overnight and then filtered through celite, eluting with dichloromethane. The solvent was removed in vacuo and the resulting residue dissolved in 10 mL of dichloromethane. The dichloromethane solution was washed with 10 mL of 1 M NaOH and 10 mL of brine. The organic phase was dried over MgSO4, and the solvent evaporated, resulting in 108 mg (95%) of [4-13C, 3,5-2H2]4-fluoroaniline 5 as a brown oil. 1H NMR (400 MHz, CDCl3) δ: 6.63 (dd JCH =10.5 Hz, JHF = 4.4 Hz), 3.5 (bs, 2H, NH2). 13C NMR (100.6 MHz, CDCl3) δ: 157.23 (JCF = 257.9 Hz).
[4-13C, 2,3,5,6-2H4]4-fluoroaniline 6
In a microwave vessel, 114 mg of [4-13C, 3,5-2H2] 4-fluoroaniline 5 (1 mmol) was mixed with D2O (1.5 mL) and DCl 7 N (0.5 mL). The vessel was irradiated for 2 h at 180 °C. After irradiation, 10 mL of 1 M NaOH were added, and the mixture extracted with dichloromethane (3 × 60 mL). The combined organic phases were dried over MgSO4 and the solvent was evaporated, yielding 110 mg (95%) of [4-13C, 2,3,5,6-2H4]4-fluoroaniline 6 as a pale brown oil. 1H-NMR spectroscopy indicated almost quantitative aryl deuteration with residual protons in position 2 and 3 of < 5%. 1H NMR (400 MHz, CDCl3) δ: 3.53 (bs, 2H, NH2). 13C NMR (100.6 MHz, CDCl3) δ: 157.23 (JCF = 257.9 Hz).
[1-13C, 2,3,5,6-2H4]1-fluoro-4-iodobenzene 7
The procedure was modified from literature (Madden et al. 2019). 116 mg of [4-13C, 2,3,5,6-2H4]4-fluoroaniline 6 (1 mmol) were dissolved in a mixture of 1.3 mL H2O and 0.3 mL conc. HCl in a 10 mL round-bottom flask, which was placed in an ice bath at 0 °C. 140 mg of NaNO2 in 2 mL of H2O were slowly added while maintaining the temperature below 5 °C. After completing the addition, the mixture was stirred for 20 min at 0–5 °C. Next, 332 mg of NaI (2 mmol) in 2 mL of H2O were added gradually over 20 min. The mixture was stirred at room temperature for 30 min before heating to reflux for another 30 min. After cooling to room temperature, the solution was neutralized with 1 M NaOH, and then 30 mL of 10% Na2S2O3 solution were added. The aqueous phase was extracted with dichloromethane (3 × 50 mL), and the combined organic layers were dried over MgSO4. The solvent was evaporated, yielding a pale-yellow oil, which was further purified by bulb-to-bulb distillation at 90 °C and 20 mbar, resulting in 172 mg (76%) of [1-13C, 2,3,5,6-2H2]1-fluoro-4-iodobenzene 7 as a colorless oil. 13C NMR (100.6 MHz, CDCl3) δ: 162.84 (d, JCF = 247.0 Hz, 13CF).
[4-13C, 2,3,5,6-2H4]
N-[(1,1-dimethylethoxy)carbonyl]-4-fluoro-l-phenylalanine methyl ester 8
Zinc dust (190 mg, 3 mmol) was loaded to a flame dried, argon purged round-bottom flask. Dry DMF (1 mL) was added via syringe followed by a catalytic amount of iodine (40 mg, 0.15 mmol). N-Boc-3-iodo-l-alanine-methylester (329 mg, 1 mmol) was added immediately followed by a catalytic amount of iodine (40 mg, 0.15 mmol), which resulted in a significant rise of temperature. After the solution had come to room temperature again, Pd2dba3 (22 mg, 0.025 mmol), SPhos (21 mg, 0.05 mmol) and 295 mg of [1-13C, 2,3,5,6-2H4]1-fluoro-4-iodobenzene 7 (1.3 mmol) were added to the flask and left to stir at room temperature for 24 h under argon. The crude reaction mixture was purified using silica gel column chromatography to yield 272 mg (90%) of product 8 as a brown solid. 1H NMR (400 MHz, CDCl3) δ: 1.42 (9 H, s), 3.01 (dd, 1H, J = 13.9 Hz and 5.6 Hz), 3.11 (dd, 1H, J = 13.9 Hz and 6.0 Hz), 3.72 (s, 3 H), 4.57 (m, 1H), 4.97 (d, 1H, J = 8.4 Hz). 13C NMR (100.6 MHz, CDCl3) δ: 161.96 (d, JCF = 245.2 Hz, 13CF). HRMS (ESI) for C14H16O413CD4FN; m/z = 325.1550 ([M + Na]+calc. = 325.1553).
[4-13C, 2,3,5,6-2H4]4-fluoro-l-phenylalanine 9
ln a 50 mL round bottom flask, 302 mg of [4-13C, 2,3,5,6-2H4]N-[(1,1-dimethylethoxy)carbonyl]-4-fluoro-l-phenylalanine methyl ester 8 were dissolved in 7 mL of MeOH, treated with a solution of LiOH·H2O (145 mg in 2.5 mL of water) at RT and stirred overnight. Then, the mixture was washed with 2 × 10 mL of diethyl ether. The aqueous layer was acidified with 1 M HCl to pH 2 and extracted with 4 × 15 mL of ethyl acetate. The combined organic phases were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The resulting viscous oil was taken up in 5 mL of dioxane under argon and a mixture of cold 4 M HCl (9 mL) and dioxane (5 mL) was added dropwise over 10 min. at 0 °C. The reaction was stirred for another 10 min at this temperature, then allowed to warm to room temperature and stirred for another 2 h. Dioxane was removed using a rotary evaporator and the resulting solid triturated with 40 mL of diethyl ether and few drops of ethyl acetate. The resulting solid was isolated by filtration to give 220 mg (98%) of product 9. 1H NMR (400 MHz, D2O) δ: 3.19 (dd, 1H, J = 14.5 Hz and 7.5 Hz), 3.32 (dd, 1H, J = 14.5 Hz and 5.5 Hz), 4.18 (dd, 1H, J = 7.5 Hz and 5.5 Hz). 13C NMR (100.6 MHz, H2O) δ: 162.08 (d, JCF = 243.5 Hz, 13CF). HRMS (ESI) for C8H7O213CD4FN; m/z = 189.1049 ([M + H]+calc. = 189.1053).
Expression and purification of 19F/13C/2H phe GB1
His6-TEV-GB12−56 (MKHHHHHHPM SDYDIPTTEN LYFQ/GAMA QYKLILNGC TLKGETTTEA VDAATAEKVF KQYANDNGVD GEWTYDDATK TFTVTE) was expressed in BL21(DE3) phage resistant E.coli using a pETM-11 vector system. Cultures were grown in a 15N-enriched M9 minimal medium (1 g/L 15NH4Cl) at 37 °C under agitation (140 rpm) until OD600 had reached 0.7. Then 2 g/L glyphosate along with 120 mg/L L-Trp and 120 mg/L L-Tyr and 100/200/400 mg/L (sample 1, 2 and 3) of [4-13C, 2,3,5,6-2H4] 4-fluorophenylalanine 9 were added. After 40 min at 37 °C the temperature was lowered to 28 °C, and after another 15 min cell expression was induced by adding isopropyl-β-D-thiogalactopyranoside (IPTG; 0.4 mM final concentration). The expression culture was incubated for 16 h at 28 °C. Cells were pelleted by centrifugation, resuspended in PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) and lysed by sonication. After centrifugation the supernatant was filtered and purified with a HisTrap FF crude column (GE Life Sciences) using an imidazole gradient (500 mM). The buffer was exchanged to a TEV-cleavage buffer (50 mM Tris-HCl, 0.5 mM EDTA, 1 mM DTT, pH 8) and the protein incubated with TEV protease overnight at 4 °C. The protein was further purified via a reversed HisTrap step. All NMR measurements were performed in a 50mM NaAc buffer (1 µM NaN3, + 1 mM fresh DTT, pH 5.5). Non-19F-labeled GB1 (sample 0) was expressed in the same way, without addition of glyphosate, Trp, Tyr and compound 9. The protein yields for samples 1–3 in 100 mL M9 minimal medium cultures each have been determined to 0.39 mg, 0.32 mg and 0.16 mg, respectively. This corresponds to 500 µl samples with concentrations of 0.12, 0.1 and 0.05 mM.
NMR experiments
2D 15N-1H spectra, employing the SOFAST HMQC NMR pulse sequence (sfhmqcf2gpph; d1 = 0.2 s, aq time = 0.06 s, 384 points the i.d. (0.1 s), sw of 15.63 ppm (1H) × 35 ppm (15N), garp 13C-decoupling, and, depending on the sample concentration, 4–32 scans) (Schanda et al. 2005), and 1D 19F spectra (zgig; d1 = 1 s, aq time = 1.0 s, sw of 40 ppm, 8192 scans and waltz16 1H-decoupling scheme) were both acquired at 298 K on a Bruker® Avance Neo 500 spectrometer (11.7 T), using a broadband BBO probe.
1D 19F NMR (zgig; d1 = 1 s, aq time = 0.5 s, sw of 30 ppm, 1024 scans) and 2D 13C -19F HSQC (hsqcetgpsi, d1 = 1.5 s, aq time = 0.025 s, 128 points in the i.d. (0.012 s), sw 30ppm (19F) × 30ppm (13C), 128 scans, garp 13C-decoupling) spectra were acquired at 298 K on a Bruker® AV III HD + 700 MHz spectrometer (16.4 T) using a quadruple-resonance QCI_F (1H, 13C, 15N, 19F) helium cooled cryo-probe. The 19F chemical shifts are referenced to KF (δ = −125.3 ppm), which was specifically co-resolved in the sample for referencing. NMR data was processed and analyzed using TopSpin® software.


留言 (0)