
記住我

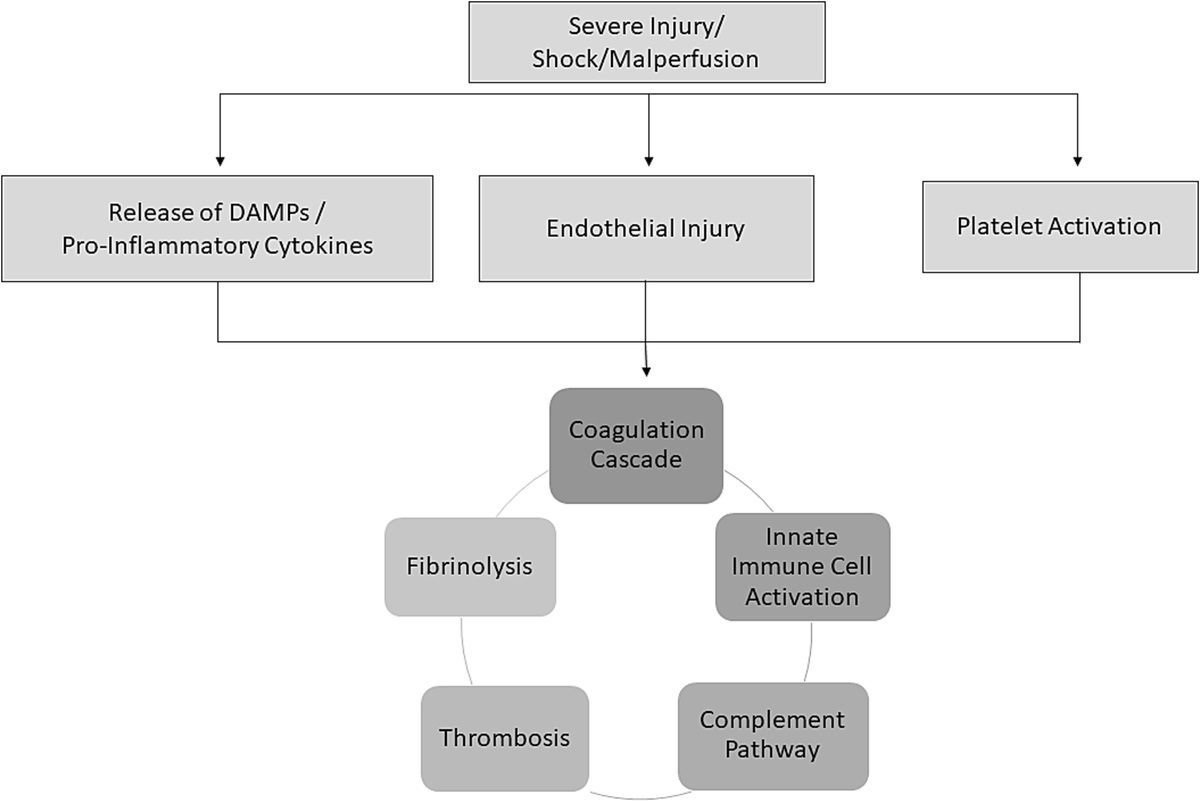

Coagulopathy and acute organ dysfunction are indicative of the systemic inflammatory response syndrome (SIRS) that is frequently seen in severely injured patients. While we often note these clinical signs in patients during the early postinjury management, these events do not occur in isolation. In fact, the coagulation system, immune system, and complement system all interact and precipitate coagulopathy, platelet dysfunction, cytokine release, immune cell activation, and endotheliopathy that must be resolved to ensure adequate resuscitation and recovery from injury (Fig. 1). Knowledge of the extensive crosstalk that occurs between these systems is essential to understanding the physiology of severely injured patients and to optimize the return to homeostasis through resuscitation and correction of coagulopathy. Promptly correcting coagulopathy and resolving SIRS is essential to mitigate organ dysfunction and multiorgan failure that can cause significant morbidity postinjury.
 Figure 1:
Figure 1: Diagram demonstrating the interconnected components of the systemic inflammatory response to injury.
THE SYSTEMIC INFLAMMATORY RESPONSE TO INJURYSystemic inflammatory response syndrome occurs early after injury and is an essential immune response to tissue injury, although it may also be deleterious to the host. Clinically, SIRS is defined as two or more of the following criteria: temperature of >38°C or <36°C, heart rate of >90 beats per minute, white blood cell count of >12,000 or <4,000 per microliter or >10% bands, respiratory rate of >20 breaths per minute, or pCO2 level of <32 mm Hg. Underlying these clinical alterations, a host response includes activation of innate and adaptive immune cells, cellular and noncellular coagulation cascades, and complement systems, resulting in a widespread and interrelated inflammatory response. Characterization of the inflammatory response to major injury reveals a rapid and dramatic change in the expression of genes related to innate and adaptive immunity. The evaluation of the leukocyte transcriptome from the Inflammation and Host Response to Injury program demonstrated more than 5,000 genes with at least twofold change in expression in patients after major injury, with the greatest changes occurring less than 12 hours from the time of injury.1 Interestingly, there was a simultaneous increase in genes related to innate immunity, with suppression of genes related to adaptive immunity. These leukocyte genomic changes persisted for longer in patients with complicated recovery from injury, suggesting that resolution of the SIRS response is a key factor in recovery. In addition to dramatic changes in the immune system, there are multiple other host changes after injury, including activation of endothelial cells and platelets, as well as alterations to the coagulation system and complement cascade. The interaction of these systems contributes to thromboinflammation in severely injured patients and must be understood to optimally address resuscitation and mitigate trauma-induced coagulopathy (TIC) and its complications.
THE INNATE IMMUNE SYSTEMThe innate immune system provides early defense against injury by recognizing damage-associated molecular patterns (DAMPs) released by injured cells and damaged tissue, leading to cytokine release, further activation and recruitment of immune cells, and further amplification of the inflammatory response.2 Neutrophils play a major role in the early immune response to injury and are early effectors of the inflammation.3 Under proinflammatory conditions, neutrophils demonstrate significant interaction with the endothelium, platelets, and the coagulation processes, including through the formation of neutrophil extracellular traps (NETs). Neutrophil extracellular traps are networks of extracellular fibers that allow neutrophils to kill pathogens while minimizing oxidative burst-induced damage to other cells. Recent studies on the inflammatory response to injury indicated that NETosis correlates with increasing injury severity.4–7 The role(s) of leukocytes and NETs in inflammation and thrombosis remain an area of ongoing investigation. Still, their regulation may eventually yield strategies to therapeutically manipulate thromboinflammation after injury.8 Neutrophils also migrate to sites of tissue injury via the extracellular mask tricks, where they degranulate through oxidative burst and cause cytotoxicity. While required to control local tissue injury, this can also damage host cells. Because neutrophils are early responders, secretion of cytokines, including tumor necrosis factor (TNF) and interferon γ, is necessary to further recruit other innate immune cells.
Neutrophils and monocytes are released from the bone marrow into the circulation in response to injury, in a process known as emergency myelopoiesis.9 Monocytes also play an important role in the early innate immune response by patrolling blood vessel walls, interacting with the endothelium, regulating transendothelial migration, and propagating the immune response through cytokine release. They also mature into macrophages after leaving the circulation, playing an important role in the phagocytosis of cellular debris. A recent study of peripheral blood drawn from severely injured patients within the first 12 hours postinjury demonstrated phenotypic and functional changes in circulating neutrophil and monocyte subsets compared with blood collected from healthy volunteers.10 This included increased phagocytic activity and increased production of reactive oxygen species, further supporting dysregulation of the innate immune response following trauma. Not only does immune cell recruitment and cytokine release amplify the innate immune response, but it also interacts with the coagulation system to further drive thromboinflammation after injury.
THE INTERSECTION OF COAGULATION AND INFLAMMATIONThe study of TIC has been long appreciated as pivotal for advancing the science behind hemorrhage control for injured patients.11,12 However, the intersection of TIC with inflammation and immune responses has yet to receive equal focus in efforts to improve outcomes after injury.13 In thromboinflammation, coagulation cascades intersect with inflammatory pathways through various mechanisms and cellular interactions (Fig. 2). Coagulation factors, especially tissue factor (TF) and factor VIIa, interact with immune cells, triggering the release of inflammatory mediators and amplifying inflammation.14 Reciprocally, inflammatory mediators can also activate coagulation by promoting the expression of TF on endothelial cells and monocytes. Severe injury is associated with the release of proinflammatory mediators, which alter the circulatory system's normal ratio of procoagulation and anticoagulation effects. The overall proinflammatory response after injury leads to a dysregulated balance, with endothelial cell, platelet, and leukocyte activation that results in appropriate and inappropriate thrombus formation, thrombolysis, and exhaustion of clotting cycle components (Table 1).
 Figure 2: The intersection of inflammation, coagulation, and thrombosis during SIRS. Proinflammatory cytokines and DAMPs propagate the inflammatory response to innate immune cells, the endothelium, and the coagulation system. Polymorphonuclear leukocytes are activated and can form NETs at the surface of the activated endothelium. Endothelial injury can expose the underlying subendothelium exposing TF and collagen, which can activate the clotting cascade. Activated platelets play an important role in this inflammatory response where they can cause thrombosis in association with fibrin and vWF at the endothelial surface. Systemic inflammatory response syndrome also causes shedding of the endothelial glycocalyx and activation of fibrinolytic pathways. Diagram was created with Biorender.com.
TABLE 1 -
Selected Key Mediators of the Intersection of Coagulation and Inflammation
Selected Mediator
Source
Role in Thromboinflammation
Activated protein C
Plasma
Natural anticoagulant with cytoprotective role; promotes endothelial barrier; inhibits proinflammatory mediators
Complement cascade
Plasma
Stimulates platelet activation and adhesion
DAMPs
Injured cells
Cytokine release and inflammatory response amplification
Elastase
Neutrophils
Decreased circulating antithrombin
EVs
Multiple
Propagates inflammatory response
Factor VIIa
Plasma
Triggers release of inflammatory mediators from immune cells
Fibrin
Plasma
Promotes leukocyte adhesion, migration, and cytokine release
P-selectin
Platelets
Recruits immune cells; induces cytokine release; receptor for C3b, initiates complement activation
Phosphatidylserine
Platelets
Recruits immune cells; induces cytokine release
Platelet factor 4
Platelets
Recruitment and activation of neutrophils and monocytes
Cytokines
Multiple
Increased TF expression on endothelial cells and monocytes
Thrombin
Plasma
Cleaves protease-activated receptors leading to cytokine release
TF
Multiple
Triggers release of inflammatory mediators
Toll like receptors
Platelets
Recognizes PAMPs and DAMPs, triggers inflammatory responses
vWF
Multiple
Propagates platelet adhesion
Figure 2: The intersection of inflammation, coagulation, and thrombosis during SIRS. Proinflammatory cytokines and DAMPs propagate the inflammatory response to innate immune cells, the endothelium, and the coagulation system. Polymorphonuclear leukocytes are activated and can form NETs at the surface of the activated endothelium. Endothelial injury can expose the underlying subendothelium exposing TF and collagen, which can activate the clotting cascade. Activated platelets play an important role in this inflammatory response where they can cause thrombosis in association with fibrin and vWF at the endothelial surface. Systemic inflammatory response syndrome also causes shedding of the endothelial glycocalyx and activation of fibrinolytic pathways. Diagram was created with Biorender.com.
TABLE 1 -
Selected Key Mediators of the Intersection of Coagulation and Inflammation
Selected Mediator
Source
Role in Thromboinflammation
Activated protein C
Plasma
Natural anticoagulant with cytoprotective role; promotes endothelial barrier; inhibits proinflammatory mediators
Complement cascade
Plasma
Stimulates platelet activation and adhesion
DAMPs
Injured cells
Cytokine release and inflammatory response amplification
Elastase
Neutrophils
Decreased circulating antithrombin
EVs
Multiple
Propagates inflammatory response
Factor VIIa
Plasma
Triggers release of inflammatory mediators from immune cells
Fibrin
Plasma
Promotes leukocyte adhesion, migration, and cytokine release
P-selectin
Platelets
Recruits immune cells; induces cytokine release; receptor for C3b, initiates complement activation
Phosphatidylserine
Platelets
Recruits immune cells; induces cytokine release
Platelet factor 4
Platelets
Recruitment and activation of neutrophils and monocytes
Cytokines
Multiple
Increased TF expression on endothelial cells and monocytes
Thrombin
Plasma
Cleaves protease-activated receptors leading to cytokine release
TF
Multiple
Triggers release of inflammatory mediators
Toll like receptors
Platelets
Recognizes PAMPs and DAMPs, triggers inflammatory responses
vWF
Multiple
Propagates platelet adhesion
PAMP, pathogen-associated molecular pattern.
Coagulation factors, particularly thrombin, have been shown to modulate immune responses because thrombin can cleave protease-activated receptors on endothelial cells, immune cells, and platelets, leading to the release of cytokines, chemokines, and adhesion molecules.15 Furthermore, fibrinogen and fibrin play multifaceted roles influencing both thrombosis and inflammation, with effects that extend beyond serving as a passive substrate for clot formation. Fibrin can activate immune cells through integrin receptors, promoting leukocyte adhesion, migration, and cytokine release, and provides a scaffold for the assembly of immune cells and facilitation of neutrophil and monocyte activation and function.16 Moreover, fibrin complexes can serve as a platform for the deposition of complement components, amplifying inflammatory responses, sequestering cytokines and growth factors, and releasing them upon fibrinolysis.17 Increased plasminogen activator inhibitor 1 (PAI-1) levels can impair the fibrinolytic system, leading to a prothrombotic environment. This imbalance contributes to thrombosis and sustains inflammation by preventing timely fibrin degradation.
The concept of fibrinolysis shutdown, described in injury, is also seen in prothrombotic and proinflammatory infectious diseases, including coronavirus disease 2019 (COVID-19). Death in patients with fibrinolytic shutdown after injury has been associated with organ failure,18,19 presumed to be related to microvascular thromboses and the relationship of the imbalanced fibrinolytic system with thromboinflammation. Furthermore, it has been found that patients with persistently low fibrinolytic activity after injury have an increase in later mortality,20,21 likely because of persistently high PAI-122 and neutrophil elastase suppression of fibrinolysis.23 The fibrinolytic system is also under complex and bidirectional regulatory control by platelets. For example, the tissue plasminogen activator and plasminogen binding capacity of platelets play a role in activating fibrinolysis. Still, the platelet-induced contraction of fibrin-rich clots and the platelet release of PAI-1–rich α granules are antifibrinolytic in nature.24,25 The extensive role of platelets in thromboinflammation is discussed further hereinafter.
Antithrombin is the main natural inhibitor of factor Xa and thrombin and can also be altered in the setting of posttraumatic SIRS. Activation of proinflammatory pathways may decrease circulating antithrombin levels through degradation by elastase from activated neutrophils, decreased synthesis, and consumption.26 Antithrombin itself may also play a role in the inflammatory response by indirectly suppressing platelet activation, decreasing leukocyte rolling on endothelial cells, and suppressing the production of proinflammatory cytokines.26 Recent clinical data demonstrate that injury decreases plasma antithrombin levels.27 Given the overall anti-inflammatory role of antithrombin, the decrease in antithrombin seen after injury may predispose patients to worsened systemic inflammation and a prothrombotic state.
ROLE OF PLATELETS IN THROMBOINFLAMMATIONPlatelets are anucleate blood cells originating from megakaryocytes in the bone marrow. Classically, their “primary” biological responsibility has been linked to hemostatic control.28 Endothelial injury and tissue damage expose subendothelial matrix components, including TF, von Willebrand factor (vWF), and collagen, triggering platelet activation and providing activated platelet surfaces for coagulation factor assembly.29 The ligand interaction of platelet receptor glycoprotein Ibα with vWF leads to the recruitment of platelets to sites of vascular injury. Platelets rapidly adhere to the exposed subendothelial matrix, promoting primary hemostasis. In addition, platelet activation promotes the exposure of phosphatidylserine, a key component in the amplification of coagulation. The procoagulant phospholipid surface of activated platelets facilitates the assembly of the tenase and prothrombinase complexes, enabling the critical burst of thrombin and ultimately leading to fibrin generation. In addition, activated platelets release bioactive molecules, including thromboxane A2, adenosine diphosphate, and PAI-1, which further enhance platelet aggregation. They are involved in multiple cell-cell signaling mechanisms and regulation of the microvascular environment.25,30
In the setting of injury and shock, platelet hemostatic functions are altered and have been the subject of much research related to TIC.31,32 It remains incompletely understood whether described platelet alterations are maladaptive biology or evidence of adaptive functions in the setting of hypoperfusion and hemorrhage.33,34 Furthermore, evidence highlights that platelets play a crucial role in vascular biology beyond hemostasis.35 The activation of platelets and complex interplay of receptors and signaling molecules mediates thromboinflammation through the vWF axis,36 platelet shape and receptor conformation changes,37 secretion of granule contents,25,30 and exposure of procoagulant surfaces.28 Activated platelets express phosphatidylserine and P-selectin, which not only allow for coagulation factor binding and thrombin generation, but these procoagulant platelets signal through both the release of soluble contents and direct contact with leukocytes and endothelial cells, recruiting immune cells to local environments and inducing their release of cytokines and chemokines that promote inflammation.38,39
The release of platelet factor 4 is associated with proinflammatory states of increased expression of TF, levels of fibrinogen, and activated factor X.40 Platelet-derived molecules such as platelet factor 4 and β-thromboglobulin can stimulate immune cells, contributing to the recruitment and activation of neutrophils and monocytes, and NET formation. In addition, other proinflammatory platelet phenotypes have been identified after injury, including histone H4 decorated procoagulant ballooning, as well as platelet-derived soluble CD40 ligand and platelet-derived high mobility group box 1.41,42 Furthermore, platelets play a sentinel role in immune surveillance through their expression of toll-like receptors that recognize pathogen-associated molecular patterns and DAMPs, triggering inflammatory responses. In fact, the platelet signalosome includes the expression of all 10 toll-like receptors transcripts43 and a plethora of other immune receptors for complement, antibodies, and intracellular nucleotide-binding and oligomerization domain–like receptors.44
The interplay between activated platelets, coagulation factors, and immune cells creates a localized environment primed for thrombosis and inflammation, which can lead to microvascular dysfunction, tissue edema, and injury exacerbation. It is likely that the known alterations to platelet function after injury have downstream effects on the regulation of endothelial and immune functions and contribute to the microvascular thrombotic complications seen in injured patients who develop venous thromboembolism and multiple organ failure.42 In addition, in other diseases that are also characterized by ischemia-reperfusion injury, it has been identified that platelets carry a signaling role in the regulatory T cell suppression.45 Finally, platelets and endothelial cells are symbiotic in their joint regulation of hemostatic functions and inflammatory pathophysiology.46,47
ROLE OF ENDOTHELIUM IN THROMBOINFLAMMATIONEndothelial activation is a central event in thromboinflammation.48 The endothelium, a monolayer of cells lining blood vessels, is a critical interface between circulating blood and underlying tissues, serving as a nexus of both hemostasis and inflammation. Beyond its well-appreciated role as a dynamic barrier regulating vascular tone and permeability, the endothelium also plays a governing role in noncellular and cellular signaling across the vascular space. Under normal conditions, endothelial cells discourage clot formation through the interaction of heparan sulfate with circulating antithrombin, forming a functional layer of anticoagulant at the endothelial cell surface. In pathological conditions, such as injury, the endothelium undergoes phenotypic changes that promote thrombosis and inflammation, contributing to microvascular clot formation, tissue damage, and immune cell recruitment.
Injury-induced endothelial dysfunction, often termed endotheliopathy of trauma, has been increasingly appreciated for its role in TIC and associated complications.49–51 Dysfunctional endothelium involves the shedding of the proteoglycan layer and a denuded loss of barrier function but also promotes thrombus formation and allows immune cells to infiltrate tissues, intensifying the inflammatory response.52 Multiple described pathways may contribute to endotheliopathy of trauma in injury and shock, including via thrombin-thrombomodulin, protein C, and heparan sulfate biology.11,53–55 The microvascular thrombosis driven by endothelial activation and dysfunction can have profound consequences, including occlusion of the microvasculature, reducing tissue perfusion and leading to ischemia and hypoxia, as well as endothelial cell mechanical damage exacerbating vascular leakage and tissue edema. The end result of this pathophysiology is organ failure.56
Like the other mediators of thromboinflammation discussed previously, this biology does not happen in isolation, and activation and injury to the endothelium can affect the endothelial symbiosis with platelets and fibrinogen.57 This includes the role of platelets in angiogenesis and the release of soluble platelet-derived CD40, involved in endothelial and inflammatory signaling, and found to be associated with TIC and associated poor outcomes.58 In addition, the closely tied biology of vWF and its clearance by a disintegrin and metalloproteinase with thrombospondin type 1 motif, 13 (ADAMTS13), are under endothelial control, and it has been found that sustained release and impaired clearance of ultralarge vWF are associated with thromboinflammatory biology.46,59,60
The effects of malperfusion in vascular beds should not be ignored.61 Specifically, malperfusion triggers inflammation through the release of DAMPs, including extracellular nucleic acids and purinergic nucleotides, as well as high mobility group box 1 and cytokines. These alarmins are then recognized by surrounding cells in the vascular space, including the endothelial cells, immune cells, and platelets, leading to the convergence of prothrombotic proinflammatory phenotypic function of these cells.62 For the endothelial cells, this has been found to include endothelial plasticity in which endothelial cells lose endothelial characteristics and acquire mesenchymal characteristics.62 Furthermore, complement and endothelial cell activation are codependent, and the complement role in thromboinflammation is described further hereinafter.63
Tissue factor (factor III or CD142) is a cell surface glycoprotein constitutively expressed by cells not normally exposed to the circulatory system, including fibroblasts and subendothelial smooth muscle cells. Injury may lead to direct exposure of these tissues to the circulation, with binding of the serine protease factor VIIa with resultant conversion of factor X into factor Xa and subsequent conversion of prothrombin to thrombin.64 Tissue factor expression is inducible in monocytes, endothelial cells, platelets, and other organ systems through activation of proinflammatory transcription factors after traumatic injury.65 In this setting, increased TF expression leads to an overall prothrombotic state, with thrombosis at the site of endothelial cell TF expression. After injury, increased TF activation may lead to the exhaustion of proteins in the clotting cascade, leading to coagulopathy.
Activated protein C is a vitamin K–dependent serine protease that inactivates factor Va and factor VIIIa, thus serving as a natural anticoagulant. The zymogenic form of protein C is present in the circulation under baseline conditions and is activated by binding with thrombin as well as through interactions with thrombomodulin and the endothelial protein C receptors. Because of the location of these receptors in the endothelial cell membrane, activated protein C is typically found in proximity to the endothelium where it affects the blood/endothelial cell interface. Activated protein C also has a cytoprotective role, primarily through the endothelial protein C receptor and protease activated receptor 1. This effect is incompletely understood but involves promotion of the endothelial barrier, inhibition of proinflammatory mediators, and decreased expression of vascular adhesion molecules with reduction of leukocyte adhesion and chemotaxis.66 Severe injury is associated with significant early elevation of activated protein C levels, and this finding is associated with alterations in clotting factor activity, with the net result of increased coagulopathy and decreased clot strength as well as increased fibrinolysis and fibrinogen depletion. There is debate regarding whether this is a primary driving factor for the development of TIC.67,68 Recent studies also suggest that cell free histones, which are markedly elevated after injury and the ensuing inflammatory response, lead to inhibition of protein C activation.69 Previous clinical studies led to the treatment of sepsis with activated protein C. Although the potential clinical harm of this therapy was subsequently demonstrated to outweigh any potential benefits,70 recent experimental data suggest that therapeutic manipulation of the protein C could have a future role in mitigation of TIC.71
THE ROLE OF COMPLEMENT IN THROMBOINFLAMMATIONAs a major component of the innate immune response and an ancient defense mechanism, the complement system is consisted of a series of soluble and membrane-bound proteins that play a crucial role in immune surveillance and inflammation. The complement system has gained increasing recognition for its crosstalk with the coagulation system and its role in thromboinflammation. There are multiple pathways by which the complement system can interact with coagulation pathways. Complement activation can occur through classical, lectin, or alternative pathways. Activation generates anaphylatoxins (C3a, C5a), opsonins (C3b, iC3b), and the membrane attack complex (C5b-9), which influence various cellular responses.72 Anaphylatoxins can stimulate platelet activation and adhesion, promoting clot formation.73 Furthermore, lectin-pathway protease activity is triggered by platelet activation and fibrin generation, and reciprocally, these complement pathway proteases can catalyze coagulation proteins, including prothrombin, fibrinogen, factor XIII, and thrombin-activatable fibrinolysis inhibitor.74 Impressively, this is a process by which fibrin can form in the absence of thrombin.75 In addition, opsonized surfaces may enhance coagulation factor assembly and thrombin generation. Complement activation promotes inflammation by recruiting immune cells, enhancing vascular permeability, decreasing nitric oxide production, and releasing proinflammatory cytokines.76 Complement activation fragments, particularly C3a and C5a, interact with immune cells through their respective receptors, modulating leukocyte chemotaxis and activation.77 Furthermore, the membrane attack complex can directly damage cells, releasing DAMPs that fuel the inflammatory response. Complement activation results in the shedding of complement-coated endothelial microvesicles and the assembly of C5b-9, which can ultimately lead to cell lysis, and the procoagulant TF activity on the endothelium is dependent on the assembly of C5b-9.63,78
Dysregulation of complement activation has been implicated in various pathological conditions characterized by coagulopathies and inflammatory pathologies, including injury. Injury triggers a cascade of events that can activate the complement system.79 The described mechanisms are canonical and noncanonical and are still the topic of exploratory research and debate. There are data to support that there can be both local and systemic activation of complement,80 including activation of the classical pathway with alternative pathway amplification, increased ratios of C3a:C3b-9,81 C4a thrombin binding,82 changes to neutrophil complement regulatory proteins and associated function,83 and the activation of complement due to coagulation proteases.84 Similar to other coagulation and inflammation biology in severe injury and shock, hypotheses and studies suggest that exhaustion of complement function in the setting of excessive activation may put patients at risk of poor outcomes.80 Finally, like the other bidirectional biologies of thromboinflammation, various components of the complement system can also be activated by aspects of coagulation biology, including platelets, leukocytes, and endothelial cells, exacerbating the thromboinflammatory response. For example, P-selectin, expressed on activated platelets, has been identified as a receptor for C3b capable of initiating complement activation.85 This interconnected network of interactions contributes to microvascular thrombosis and tissue damage and underscores the complement system's multifunctional role beyond immune defense in multiple diseases, including injury.86
ROLE OF EXTRACELLULAR VESICLES IN MEDIATING SIRSExtracellular vesicles (EVs) are bound by lipid bilayers and released from many cell types during cellular stress.87 Extracellular vesicles include exosomes, microvesicles, and apoptotic bodies. Originally thought to be inert, it is now recognized that EVs contain proteins, lipids, metabolites, and nucleic acids from the parent cell and serve a role in cell-cell communication and inflammatory signaling.88–90 The systemic inflammatory response after injury or sepsis appears to cause EV release into the circulation,91 although the cellular source and downstream effects have yet to be defined.92,93 Activated endothelial cells also release EVs in response to various stimuli, including injury.94 These EVs carry bioactive molecules such as TF, microribonucleic acids, and cytokines, which can contribute to coagulation and inflammation by further enhancing platelet activation, promoting leukocyte adhesion, and modulating immune cell function.95 The net effect of inflammation and trauma-induced EV release on the coagulation status of the patient and their potential as a therapeutic to treat TIC is not fully understood and remains an area of ongoing investigation.
CLINICAL AND THERAPEUTIC IMPLICATIONS Bleeding/Resuscitation and Effects on SIRSClinical and preclinical studies demonstrate that hemorrhage leads to SIRS and that specific patterns of increased inflammatory markers after injury and hemorrhage are associated with decreased survival.96 The severity of the inflammatory response after hemorrhage is likely due to a profound ischemic injury during hemorrhage, followed by reperfusion injury during the resuscitation phase, leading to widespread activation of the immune system, endothelial cells, and end organs (Supplemental Digital Content, Supplementary Data 1, https://links.lww.com/TA/D397). The result is an activation of multiple inflammatory cascades, dramatically amplifying and producing a wide range of inflammatory mediators.97 Continued resuscitation may lead to recovery, whereas the continued inflammatory process may lead to multiple organ failure and progression to persistent inflammation, immunosuppression, and catabolism syndrome, with resultant prolonged chronic critical illness.98
After injury and hemorrhage, resuscitation seeks to replete lost intravascular volume and correct anemia, acidosis, and coagulopathy. The resuscitation strategy may have a profound effect on SIRS. Clinical studies demonstrate that increased use of crystalloids for resuscitation after trauma is associated with increased occurrence of hypoxemia and acute respiratory distress syndrome, suggesting that this strategy exacerbates the inflammatory response.99 The effect of blood-based resuscitation strategies on the inflammatory response after hemorrhage and resuscitation has recently been the focus of investigation. Preclinical studies indicate that, compared with Ringer's lactate, whole blood resuscitation decreases the proinflammatory response and acute lung injury.100 Importantly, studies in injured patients have supported that administering healthy plasma to injured patients may restore endothelial barrier function and reduce shed glycocalyx such as syndecan-1, presumed to be a marker of endothelial activation in injury.56,101–103
The importance of the age of stored packed red blood cells is also a controversial topic. While the transfusion of a relatively small volume of packed red blood cells does not appear to affect patient outcomes, clinical studies demonstrate increased complications and mortality in association with the use of packed red blood cells with longer periods of storage, massive transfusion, and resuscitation after injury and hemorrhage.104 Resuscitation from hemorrhage with higher storage age red blood cells is associated with neutrophil activation and lung injury in murine models.105 Additional studies suggest that increased systemic inflammation, endothelial cell activation, and microthrombi formation also occur.106,107 Efforts to blunt the effect of storage age on the inflammatory response after transfusion are ongoing and seek to maximize the quality of blood products used for resuscitation.108
Therapeutics Targeting ThromboinflammationScience in cardiovascular diseases outside of injury, accelerated by the thromboinflammatory biology of COVID-19, has created a surge of considerations in discovering and testing therapeutics targeting the coagulation-inflammation axis. Strategies aimed at modulating fibrinogen interactions with immune cells or disrupting fibrin deposition could attenuate inflammation in thromboinflammatory conditions. Anticoagulant therapies, such as heparins, have been considered for targeting hypercoagulability and modulating inflammation in thromboinflammatory diseases.109 Antiplatelet agents, such as P2Y12 inhibitors, are commonly used to prevent thrombotic events but may also impact inflammation through effects on cytokine release, platelet-leukocyte interactions, and NET formation, suggesting potential utility in modulating thromboinflammatory responses.110 Targeting platelet roles in immunity and inflammation through selectin ligand biologic antagonisms and beyond has been explored in thrombosis and inflammatory disease.111,112 Overall, targeting platelet-leukocyte interactions and platelet-derived inflammatory mediators could provide novel strategies for managing SIRS and trauma-related complications.
Similarly, understanding the role of the endothelium in thromboinflammation presents opportunities for therapeutic interventions.113 Agents that target various receptors and signaling pathways involved in endothelial activation, including anti-inflammatory drugs, adhesion molecule inhibitors, antioxidants, and endothelial protective agents, could ameliorate endothelial dysfunction in the context of injury.114 Furthermore, recent progress in mechanistic investigations of the antifibrinolytic tranexamic acid (TXA) has uncovered evidence of mitigation of endotheliopathy through various mechanisms, including enhancement of oxidative phosphorylation, inhibition of serine proteases, suppression of DAMP mitochondrial DNA, and stimulation of mitochondrial respiration.50,51 It is possible that the mortality benefit of TXA in injured patients is related to mitigation of endotheliopathy and downstream endothelial-related thromboinflammation.
Given that hyperactive complement has been associated with a detrimental role in thromboinflammation, complement inhibitors, such as monoclonal antibodies targeting C5 or its cleavage product C5a, have shown promise in ameliorating thromboinflammatory complications.86 In addition, it has been found that stored plasma is rich in multiple complement factors, including C3 and C5, and that stored platelet products contain anaphylatoxins as well.115 However, the heterogeneity of banked products and associated storage lesions continues to plague the ability to personalize the treatment of coagulation and inflammation with blood product infusion. In addition, in the setting of injury therapeutics, TXA has been shown to have effects on the complement system as well, thought to be through lysine analog direct inhibition of complement activation or indirect inhibition via its effects on plasmin and plasmin cleavage of C3 or C5.116 There remains interest in the potential for hemoadsorbance efforts to clear complement activation,117 as well as efforts in targeting specific complement types, including C1,118 C3,119 and C5.120
CONCLUSIONAn overall comprehensive understanding of thromboinflammation in the pathophysiology of trauma is required to consider clinical approaches to resuscitation and management of the injured patient (Table 2). Targeting the many interrelated components of thromboinflammation holds promise for developing therapeutic interventions that improve outcomes for injured patients (Table 3). Efforts to mitigate the acute and sustained changes in coagulation and inflammation and its complex interactions following injury could provide novel strategies for managing complications of TIC, SIRS, and multiple organ failure.
TABLE 2 - The Systemic Inflammatory Response to Injury: What You Need to Know Key Points: • The host response to injury results in a widespread and interrelated SIRS. • SIRS includes activation of innate and adaptive immune cells, cellular and noncellular coagulation cascades, and complement systems. • There is a simultaneous increase in genes related to innate immunity and suppression of genes related to adaptive immunity within 12 hours of injury. • Inflammatory mediators activate coagulation by promoting the expression of TF on endothelial cells and monocytes and alters the normal ratio of procoagulation and anticoagulation effects. • SIRS leads to endothelial cell, platelet, and leukocyte activation that result in appropriate and inappropriate thrombus formation, thrombolysis, and exhaustion of clotting cycle components. • Fibrinogen and fibrin play multifaceted roles influencing both thrombosis and inflammation, with effects that extend beyond serving as a passive substrate for clot formation by activating immune cells and promoting leukocyte adhesion and migration. • Early increases in activated protein C levels after injury increase coagulopathy, decrease clot strength, and increase fibrinolysis. • Endothelial dysfunction after injury is associated with shedding of the proteoglycan layer and loss of barrier function and promotes thrombus formation. • Activation of the complement system promotes inflammation by recruiting immune cells and enhancing vascular permeability and interacts with the coagulation system to contribute to coagulopathy.R.C. and T.W.C. contributed in the study design. R.C., T.W.C., L.Z.K., and T.P. contributed in the manuscript drafting. R.C., T.W.C., L.Z.K., and T.P. contributed in the critical revision.
DISCLOSUREConflict of Interest: Author Disclosure forms have been supplied and are provided as Supplemental Digital Content (https://links.lww.com/TA/D398).
REFERENCES 1. Xiao W, Mindrinos MN, Seok J, Cuschieri J, Cuenca AG, Gao H, et al. A genomic storm in critically injured humans. J Exp Med. 2011;208:2581–2590. 2. van Wessem KJ, Heeres M, Leliefeld PH, Koenderman L, Leenen LP. Lipopolysaccharide and hemorrhagic shock cause systemic inflammation by different mechanisms. J Trauma Acute Care Surg. 2013;74:37–43 discussion 43-4. 3. Napolitano LM, Faist E, Wichmann MW, Coimbra R. Immune dysfunction in trauma. Surg Clin North Am. 1999;79:1385–1416. 4. Goswami J, MacArthur T, Bailey K, Spears G, Kozar RA, Auton M, et al. Neutrophil extracellular trap formation and Syndecan-1 shedding are increased after trauma. Shock. 2021;56:433–439. 5. Huang MY, Lippuner C, Schiff M, Book M, Stueber F. Neutrophil extracellular trap formation during surgical procedures: a pilot study. Sci Rep. 2023;13:15217. 6. Jin J, Wang F, Tian J, Zhao X, Dong J, Wang N, et al. Neutrophil
留言 (0)