
記住我
Amyotrophic lateral sclerosis (ALS) is a multifactorial non-cell-autonomous neurodegenerative disease. Since the 1990s, several genes and mutations have been found to be involved in the pathogenesis of ALS. Based on this, several genetic animal models have been developed to study the pathology and pathogenesis of ALS, including invertebrates and vertebrates, such as yeast, flies, worms, zebrafish, mice, rats, guinea pigs, dogs, and non-human primates. Although each model displays different phenotypes, these genetic models are complementary to the pathological mechanisms of motor neuron (MNs) degeneration and the progression of ALS and are thus beneficial for research on the potential common pathogenesis of ALS through different genetic animal models, furthering the search for novel medicine treatments (Bonifacino et al., 2021).
Commonly used invertebrate genetic models mainly consist of yeast, Drosophila, and Caenorhabditis. These invertebrate experimental models were originally used to study genetics and development. These were also sometimes used to study the mechanisms associated with molecular and cell biology and are now presently propelled into the study of functional genomics and proteomics. The highly manipulable genomes of these models allow us to rapidly reproduce the transgenic lines, providing the ideal models for studying gene functions and gene and protein network interaction (Surguchov, 2021).
The full sequence of the invertebrate model genome is also easy to access for comparison with higher vertebrates and mammals in facilitating the evolution of genomic studies to rapidly produce transgenic animals by DNA transformation. Invertebrate organisms have more significant experimental advantages than their mammalian counterparts, such as a short generation time, small size, easy maintenance, and low cost. Another advantage of the invertebrate model is that it is amenable to genetic studies forward from phenotype to gene and reverse from gene to phenotype. Classic forward genetics studies of invertebrate models allow us to identify new molecules or pathways involved in special cell processes, which is a key advantage of invertebrate models. Forward genetics studies that apply a chemical mutagen to invertebrate models can generate target gene mutants to elucidate target gene functions. Reverse genetic studies using invertebrate models can rapidly identify possible molecular and biological pathways of certain target genes. The knockdown of target gene mutants using RNA interference (RNAi) technologies in invertebrate models can dramatically reduce target gene products by introducing double-stranded RNA (dsRNA) in models and provide related data about the roles of some target genes in biological processes.
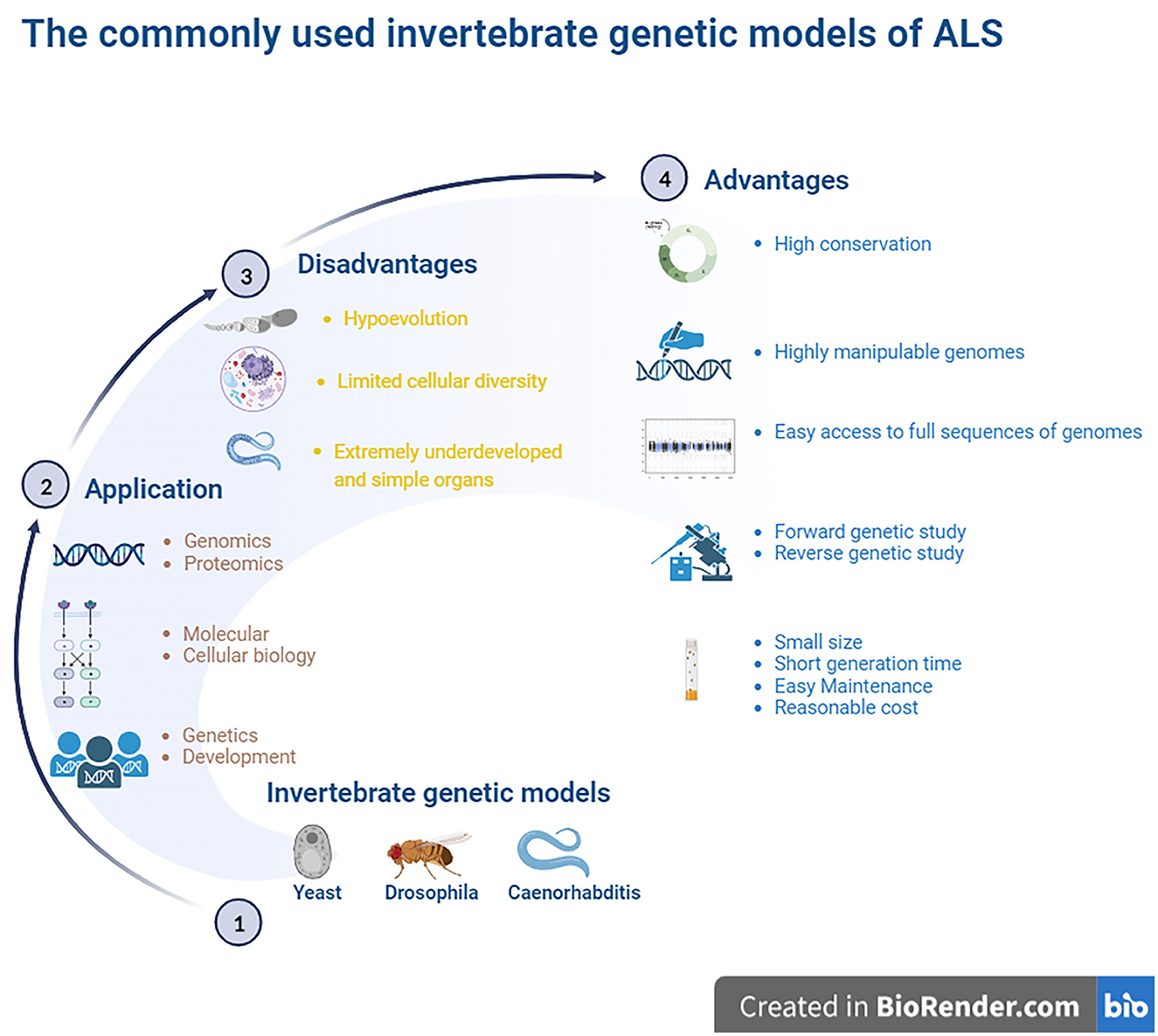
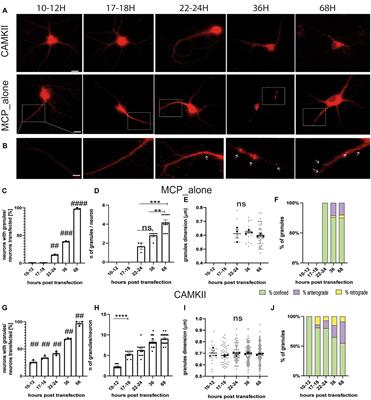
In addition to being amenable to forward and reverse genetic approaches, the advantages of invertebrate models include the following: yeast models possess a controlled environment, with a single system, an individual cell or tissue mechanism, easy gene manipulation, and easy culture. A special advantage of Drosophila is that it possesses large chromosomes, while a unique advantage of Caenorhabditis is its transparent body. Brenner (1974), Horvitz and Sulston (1980), and Sulston and Horvitz (1981) were awarded the Nobel Prize in Physiology or Medicine in 2002 for their seminal contributions concerning the genetic regulation of organ development and programmed cell death using the Caenorhabditis elegans (C. elegans) model as an investigative tool. The findings from these models established by them revealed some key genes involved in cell division, differentiation, and apoptosis and confirmed that both the structures and functions of these genes were highly conserved in higher animals, including humans. Based on their findings, they provided a framework in which simple animals can be used to identify potential molecular pathways and related biological processes. In addition, the advantages of Drosophila and Caenorhabditis also include a short life cycle, high fertility, a less complex nervous system, fully sequenced genome, which is homologous by more than 65%-75% to human genes, molecular pathways similar to humans, availability of mutagenic and transgenic techniques, and low cost (Gois et al., 2020). An important advantage of these models is that their data and information are often directly applied to research mechanisms of human diseases. Another advantage of establishing a model that imitates human disease using small invertebrate organisms is the high degree of conservation in mammalian organisms, which is useful for identifying the molecular components involved in pathogenesis. However, a major disadvantage of invertebrate models is that they are evolutionarily distant from mammals. In addition, compared to higher animals, the organs of invertebrate models are extremely undeveloped and simple, and alterations in many physiological functions and pathological and pathophysiological processes during the disease course in higher animals and mammals cannot be observed in invertebrate models. Another major disadvantage is the limited cellular diversity, which is not beneficial for studying pathological alterations at the organ and cellular levels (Gois et al., 2020) (Figure 1).
Figure 1. The commonly used invertebrate genetic models of ALS. The highly manipulative genomes of these models allow us to rapidly reproduce the transgenic lines for providing the ideal models for studying the gene functions and protein network interactions. The finds from the established models identified some key genes to involve in cell division, differentiation and apoptosis, which structures and functions were the high conservation with higher animals including humans, and it is easy access to the full sequence of model genomes. The model organisms have more significant experimental advantages, such as the short generate time, small sizes, easy maintenance as well as less cost. Besides, it is amenable to the genetic approaches of both forward and reverse. However, these invertebrate animal models are evolutionarily far from higher animal and mammalians, therefore, lots of physiologic functions can't be observed. The organs are extremely undeveloped as well as the limited cellular diversity, so it isn't beneficial to study the pathological alteration of organs and cellular levels. ALS, amyotrophic lateral sclerosis.
2 Genes involved in ALSThe majority of ALS cases are sporadic, and ~5%–10% of ALS cases are familial, with accurate Mendelian hereditary features and obvious penetrance (Gros-Louis et al., 2006). Since several missense mutations in the Cu2+/Zn2+ superoxide dismutase 1 (SOD1) gene were discovered in the subsets of familial ALS patients in 1993 (Rosen et al., 1993), various ALS-related gene mutations have been reported in the pathogenesis of ALS. Based on current genetic evidence, genetic mutations are thought to be the key cause of ALS pathogenesis. Mutations in some RNA metabolism-related genes and their related potential pathogenic mechanisms, such as disorders of cell transportation, axon outgrowth, protein metabolism, glutamatergic signaling, angiogenesis, neurotransmission, and antioxidant functions, have been confirmed to participate in the pathogenesis of ALS (Rosen et al., 1993; Neumann et al., 2006; Kwiatkowski et al., 2009; Renton et al., 2011; Morgan and Orrell, 2016). Mutations in SOD1 (Rosen et al., 1993), TAR DNA-binding protein-43 (TDP-43), fused in sarcoma/translocated in liposarcoma (FUS/TLS, FUS) (Zou et al., 2017), and open reading frame 72 on chromosome 9 (C9ORF72) genes (DeJesus-Hernandez et al., 2011) are common in major cases of ALS. In addition, several genes with less frequent mutations, such as VABP, OPTN, VCP, UBQLN2, MATR3, TBK1, NEK1, C21ORF2, ANXA11, and KIF5A are also found to participate in the pathogenesis of partial ALS patients (Leblond et al., 2014; Chia et al., 2018; Brenner and Freischmidt, 2022). Mutations in ~30 genes have already been identified in patients with ALS to date; among them, major pathological mechanisms of the known ALS genes have been identified, including SOD1, TDP43, FUS and C9orf72. Recently discovered ALS-related candidates and risk genes include SPTLC1, KANK1, CAV1, HTT, and WDR7 (Brenner and Freischmidt, 2022). To investigate the possible genetic mechanisms underlying ALS pathogenesis, various models have been established, including invertebrate models. In this review, we briefly discuss the usage, advantages, disadvantages, costs, and availability of current common invertebrate genetic models.
3 Nomenclature yeast modelsYeast models of ALS have been widely adopted to model human neurodegenerative diseases, such as ALS, since these have the advantages of easy manipulation and recapitulation compared to more complex eukaryotic cells. In particular, yeast models offer special insights into the potential relationships between the gain-of-function (GOF) toxicity of some protein aggregations. Although yeast models cannot reproduce the numerous pathogenic mechanisms involved in the degeneration of neuronal networks, they have proven to be ideal for discovering ALS-related pathogenic proteins. Among them, the yeast models of prion-like neurodegenerative disease-related proteins provide clues and valuable information for future research using a higher model system and ultimately developing therapeutics (Monahan et al., 2018).
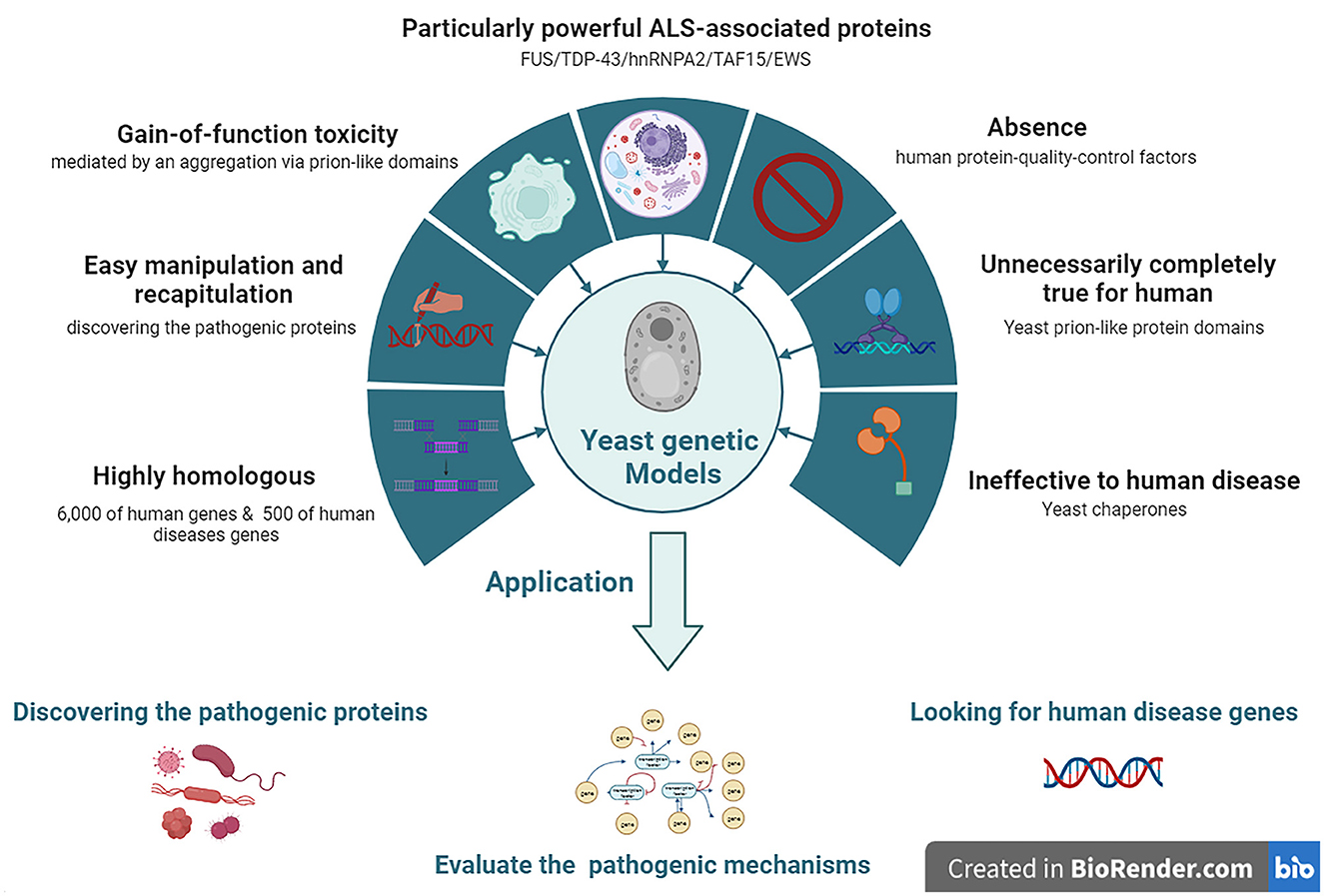
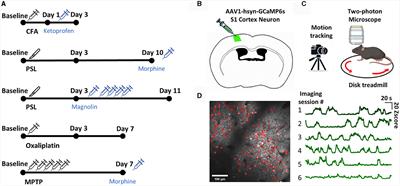
Approximately 6,000 human genes are homologous to those of yeast; among them, ~10% can be complemented with yeast (Cherry et al., 2012). Moreover, ~500 genes involved in human diseases are orthologs in yeast (Kryndushkin and Shewmaker, 2011). The most straightforward method for searching candidate pathogenic genes of human diseases among yeast is in situations where the disease genes are complemented by the yeast ortholog genes. The absence of human protein quality control factors in yeast can be an advantage in yeast models. Because of the deficiency of chaperones that regulate human pathogenic proteins among yeast, they can be useful for evaluating the chaperone-mediated changing mechanisms of aggregation and toxicity of human pathogenic proteins (De Graeve et al., 2013; Kumar et al., 2018; Park et al., 2018) (Figure 2).
Figure 2. The features and applications of Yeasts genetic models in ALS. ALS, amyotrophic lateral sclerosis; EWS, Ewing sarcoma breakpoint region 1; FUS, fused in sarcoma; hnRNPA2, heterogeneous nuclear ribonucleoprotein A2; TAF15, TATA-box binding protein associated factor 15; TDP-43, TAR DNA-binding protein 43.
3.1 Yeasts carrying SOD1 and OPTN mutationsMany neurodegenerative disease-related genes/proteins such as SOD1 and OPTN in ALS (Rabizadeh et al., 1995; Kryndushkin et al., 2012) have been established as models among yeast (Braun, 2015). To develop SOD1 yeast models, various ALS-related SOD1 mutations such as A4V, G37R, H48Q, G93A, and S134N have been introduced into the yeast SOD1 gene. The SOD1 yeast model demonstrated that the mutant human SOD1 (hmSOD1) protein is unstable and reduces cell viability but does not form insoluble SOD1 protein aggregates. Moreover, the hmSOD1 toxic effects seem not to depend on mitochondrial dysfunction or oxidative stress, but rather on the inability to control central metabolic processes, which is most probably due to the severe disruption of vacuolar compartments (Bonifacino et al., 2021).
3.2 Yeasts carrying FUS and TDP-43 mutationsALS-related proteins with prion-like domains have proven to be particularly powerful in yeast models because they are similar to naturally existing yeast prion-like proteins, such as FUS, TDP-43, heterogeneous nuclear ribonucleoprotein A2 (hnRNPA2), TATA-box binding protein associated factor 15 (TAF15), and Ewing sarcoma breakpoint region 1 (EWS) proteins, which have similar protein architectures. Moreover, these proteins were found in the neuronal cytoplasm of post-mortem ALS patients. In yeast models, ALS-related proteins and their mutant isoforms typically induce GOF toxicity, which is partially mediated by aggregation via prion-like domains. Yeast did not possess TDP-43 orthologous genes. Yeast models overexpressing human wild-type (WT) TDP-43 showed that TDP-43 accumulated and formed subcellular aggregates in the cytoplasm, which inhibited cell growth, disrupted cell morphology, and generated cytotoxicity. Yeast models of ALS-related TDP-43 mutations, such as K, M337V, Q343R, N345K, R361S, and N390D, accelerate protein aggregation, increase the number of cytosolic aggregates, and lead to growth arrest and cell death. Yeasts do not express FUS orthologs; thus, yeasts FUS models have been established by ectopically transforming human WT or mutant FUS genes into yeast. FUS yeast models are tightly associated with the translocation of FUS protein from the nucleus to the cytoplasm, forming aggregates co-localized with P bodies and stress granules in yeast cytoplasm and inhibiting the ubiquitin proteasome systems (Bonifacino et al., 2021).
Yeast models also have some limitations and disadvantages. Yeast protein domains are not the same as those in humans; thus, they must be carefully considered in studies using yeast models. The formation of prion-like proteins in yeast is its (True et al., 2004; Halfmann et al., 2012); however, this is not the case for human ALS proteins with prion-like domains. Almost all mammalian all mammalian prion-like proteins are harmful to humans (An and Harrison, 2016) since the properties of yeast prion-like protein domains are not completely conserved, but produce distinct alterations of constructs and functions that lead to contrary functions between yeast and humans during the human evolution process. Another limitation of yeast models is yeast cells do not have resident interacting proteins or chaperones that regulate human pathogenic proteins compared to mammalian cells. Chaperones are especially important for quality control and are integral to the propagation of endogenous prion proteins. Owing to the deficiency of resident interacting proteins and chaperones in yeast, the interaction of ectopically expressing human prion-like proteins in yeast is naturally different from that in humans; thus, the conclusions derived from studying protein interactions in yeast models may not be suitable for human diseases (Masison and Reidy, 2015).
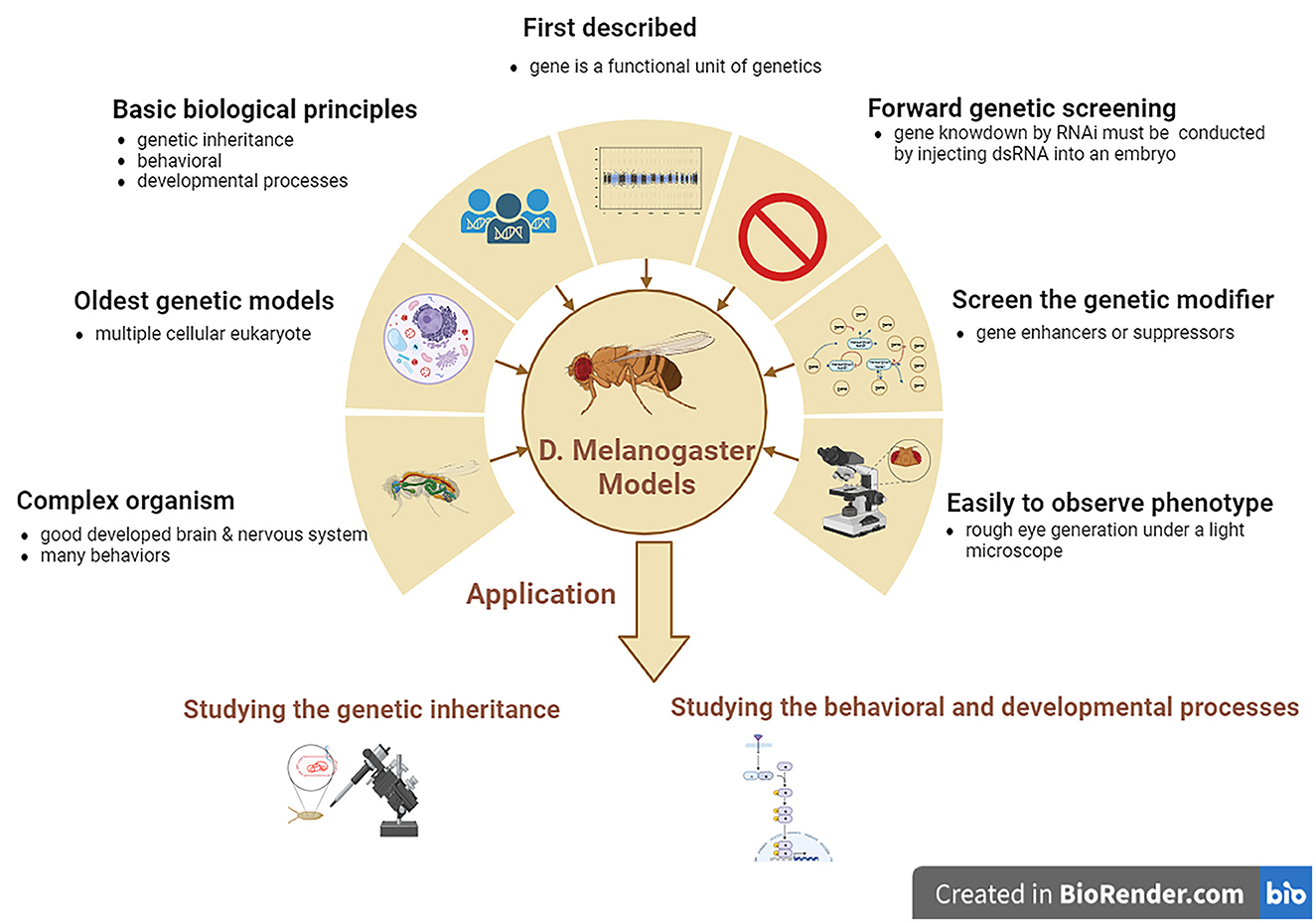
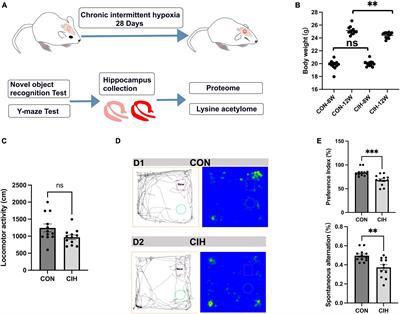
4 Drosophila melanogaster modelsDrosophila melanogaster is a good genetic model for studying neurodegenerative diseases, including ALS. D. melanogaster is a relatively complicated animal with a well-developed brain and other neural systems and display various behaviors such as learning, motor, and vision. D. melanogaster models are the earliest multiple cellular eukaryotes used as genetic models and have been applied in studying various basic biological principles, such as genetic phenotypes and genotypes, and behavioral and developed processes. The gene was first described as a functional genetic unit in D. melanogaster models. These models can be used to easily observe the neurodegenerative phenotype of rough eye generation under a light microscope. In addition, it can also be used to screen for genetic modifiers of gene enhancers and suppressors and can be used to study genetic inheritance and behavioral and developmental processes associated with human neurodegenerative diseases, including ALS. However, one main disadvantage of D. melanogaster models is that forward genetic screening by RNAi is relatively complex compared to the C. elegans model. Gene knockdown by RNAi cannot be conducted by feeding with dsRNA as simply as in C. elegans model; therefore, D. melanogaster models are made by injecting dsRNA into their embryos (Figure 3).
Figure 3. The features and applications of D. melanogaster genetic models in ALS. ALS, amyotrophic lateral sclerosis; D. melanogaster, Drosophila melanogaster.
4.1 D. melanogaster carrying SOD1 mutationsThe hSOD1 D. melanogaster models were produced by amplifying the hSOD1 gene through missense mutation technology and replacing the nitrogenous base among DNA correspondence for hSOD1 reading site with a directive mutation. The hmSOD1 D. melanogaster models exhibit movement dysfunction, local accumulation of hmSOD1 in MNs, enlargement of glial cells, neuronal degeneration, muscle contraction, decreased survival rates, and mitochondrial dysfunction (Gois et al., 2020). It reduces lifespan and fecundity and increases susceptibility to oxidative stress, movement defects, and necrotic cell death in hmSOD1 D. melanogaster models with SOD1 mutations (Phillips et al., 1989). The hmSOD1 D. melanogaster model expresses four hSOD1 mutants – G85R, H71Y, H48R and G37R. Among the endogenous D. melanogaster, SOD1 (dSOD1) produces a large amount of dSOD1 protein in neural cells (Layalle et al., 2021). Models expressing G85R, H71Y, and H48R mutations show decreased survival, developmental defects, and larval and adult dyskinesia due to muscle axon contraction (Braems et al., 2021). In addition, some antioxidant compounds have neuroprotective effects that improve exercise performance, prolong lifespan, and reduce hSOD1 cytoplasmic inclusion bodies in the hSOD1 D. melanogaster model (Liguori et al., 2021).
The transgenic D. melanogaster model expressing only hSOD1WT in MNs has been demonstrated to extend its longevity, but does not affect the movement or survival of MNs (Parkes et al., 1998). Moreover, the hSOD1WTtransgenic D. melanogaster model extended the longevity of the SOD1 deletion mutant and normal D. melanogaster but did not prevent age-dependent movement disorders. The D. melanogaster model's widespread downregulation of SOD1 expression accelerates age-dependent movement disorders and shortens the lifespan of D. melanogaster (Braems et al., 2021). Meanwhile, the D. melanogaster model selectively expressing WT or causative-related hSOD1 mutations (A4V and G85R) in MNs induces progressive movement dysfunction with electrophysiological defect, and the SOD1 aberrant accumulates stress responses around the glial cells (Watson et al., 2008). Moreover, the transgenic D. melanogaster model specifically expressing hmSOD1 in MNs showed a progressive motor protective effect, prevented the accumulation of hmSOD1 aggregation, and increased the glial cell stress response in the ventral nervous cord, accompanied by electrophysiological defects in neural circuits. The hmSOD1 transgenic D. melanogaster overexpressing mutated SOD1 can model both cellular and non-cellular autonomic damage (Walters et al., 2019). The transgenic D. melanogaster model expressing WT or mutant (A4V and G85R) hSOD1 among D. melanogaster MNs would lead to D. melanogaster climbing defects that progress over time and defects among neural circuits accompanied by the stress responses of glial cells and local accumulation of mutant SOD1 proteins among MNs (Braems et al., 2021). Meanwhile, the hSOD1 A4V transgenic D. melanogaster model showed synaptic transmission defects and focal mutation SOD1 accumulation in MNs, HSP upregulation in glial cells, cellular autonomic lesions in MNs, and abnormal alterations in glial cells. This D. melanogaster model reveals that ALS is not confined to damaging MNs, and toxic SOD1 transfers from neurons to glial cells (Clement et al., 2003; Boillée et al., 2006). The hSOD1 transgenic D. melanogaster models are usually established by hSOD1 gene expression among the MNs of D. melanogaster SOD1 null background applying GAL4/UAS yeast systems. In hSOD1 transgenic D. melanogaster models, very low levels of hSOD1WT expression are sufficient to reverse the lifespan reduction, oxidative stress increase, and physiological dysfunctions related to the SOD1 null D. melanogaster model (Mockett et al., 2003). In patients with ALS, it is very difficult to identify the complex genetics involved in its pathogenesis and to functionally investigate new candidate disease genes. D. melanogaster models can allow for an in-depth study of ALS progression from the earliest signs to terminal stages. They can rapidly perform a large range of simple assays from lifespan and motor assays to anatomical screens and can provide important functional information on the potential machinery required for proper motor neuron function and how this machinery may be dysregulated during ALS, which would not be possible in human neural systems. The D. melanogaster model is useful for addressing complex genetic diseases such as ALS. The UAS/Gal4/Gal80 system can upregulate and knock down D. melanogaster genes, including SOD1, and the ectopic expression of human SOD1 genes or mutations in a tissue-specific manner, exhibiting the typical pathologies of hmSOD1 ALS, which is beneficial for further investigation to identify disease-modifying genes, mutations, and disease pathways (Walters et al., 2019).
4.2 D. melanogaster carrying TDP-43 mutationsTDP-43 is a highly conserved and ubiquitously expressed nuclear protein that participates in various cellular processes, including mRNA splicing, transcription, stability, and transportation (Langellotti et al., 2018; Lembke et al., 2019). TDP-43 is crucial for promoting the formation and growth of neuromuscular junctions (NMJ) (Strah et al., 2020). A number of D. melanogaster models generated by the TDP-43 toxicity of endogenous D. melanogaster TDP-43 (dTDP) and hTDP-43 transgenic expression revealed that the TDP-43 protein showed toxicity, and the major phenotypes were largely similar in these D. melanogaster models. The D. melanogaster model lacking dTDP appears externally normal, but presents a deficiency in locomotion behaviors, a reduction in lifespan, anatomical defects of NMJ, and a decrease in dendrite branches (Feiguin et al., 2009; Lu et al., 2009; Lin et al., 2011). Among the above-mentioned TDP-43 D. melanogaster models, these phenotypes can be recovered and repaired by expressing the normal hTDP-43 protein in neurons, such as MNs (Feiguin et al., 2009). The phenotypes of TDP-43 D. melanogaster models revealed that the dysfunction of TDP-43 might result in the pathogenesis of ALS. In addition, overexpression of either dTDP or hTDP-43 in D. melanogaster also displayed the main pathological characteristics of ALS, premature lethality, neuronal loss, and defects in NMJ architecture and movement. TDP-43 D. melanogaster models revealed that the mechanisms of toxic GOF are related to TDP-43 proteinopathy (Lu et al., 2009; Elden et al., 2010; Hanson et al., 2010; Li et al., 2010; Ritson et al., 2010; Voigt et al., 2010; Estes et al., 2011; Miguel et al., 2011).
To study and identify the possible roles and potential pathogenic mechanisms of the TDP-43 protein in the pathogenesis of ALS, several TDP-43 transgenic D. melanogaster models have been developed to further explore whether gene modification of TDP-43 can increase or decrease the generation of TDP-43 toxicity (Elden et al., 2010; Hanson et al., 2010; Ritson et al., 2010). In these TDP-43 transgenic D. melanogaster models, upregulation of the human ATXN2 gene ortholog Pab1-binding protein 1 enhanced TDP-43 toxicity, resulting in more severe TDP-43-induced phenotypes (Elden et al., 2010). Moreover, the TDP-43 interacting partner ubiquilin 1 overexpression also produces similar TDP-43-induced phenotypes (Kim et al., 2009), reduced steady expression of TDP-43, and enhanced TDP-43 phenotypes (Hanson et al., 2010). The TDP-43-induced phenotypes among TDP-43 transgenic D. melanogaster models can also be modulated by co-expressing an ATPase member, valosin-containing protein (VCP), which is related to the protein family members of cellular activities that regulate many cell processes (Ritson et al., 2010). The above-described TDP-43 transgenic D. melanogaster models may enable the development of novel therapeutic targets that regulate TDP-43 expression in TDP-43-associated ALS patients, which is expected to prevent or treat TDP-43-associated ALS patients.
Bis-(2-ethylhexyl)-2,3,4,5-tetrabromophthalate (TBPH), a D. melanogaster TDP-43 homolog, promotes target gene transcription by combining with sequences rich in uridine guanine, thus stabilizing the binding of chromatin regulators to DNA (Zhang et al., 2018a). Overexpression of mutant or WT hTDP-43 and TBPH in transgenic D. melanogaster models affects lifespan, mobility, axonal transport, and pupal shell sealing, whereas TBPH depletion leads to movement disorders and a shortened lifespan (Walters et al., 2019; Liguori et al., 2021). Among them, hTDP-43WT overexpression in neurons results in an increase in NMJ buttons and branches associated with hTDP-43WT protein aggregation (Kankel et al., 2020). However, the phenotypes induced by the mutant hTDP-43 were not distinguishable from the hTDP-43WT induced phenotypes (Feldman et al., 2022).
Increased TDP-43 expression in TDP-43 transgenic D. melanogaster models induces mitochondrial dysfunction, including ridge abnormality and loss, resulting in a decrease in mitochondrial membrane potential and an increase in reactive oxygen species (ROS) production, usually accompanied by the loss of striated muscle tissue, and affects the survival and function of neurons due to excessive ROS production. Moreover, the TDP-43 expression increase also inhibited mitochondrial complex I activation and reduced mitochondrial ATP synthesis. Compared with the D. melanogaster control model, the volume of mitochondria in the eyes of D. melanogaster expressing TDP-43WT or TDP−43A315T was significantly reduced. TDP-43WT and TDP-43A315TGOF induced mitochondrial unfolded protein responses, including Lon and LonP1. LonP1 is a major mitochondrial matrix protease that interacts with TDP-43 to reduce mitochondrial TDP-43 protein levels, which can prevent mitochondrial damage and neurodegeneration in the D. melanogaster model induced by TDP-43 (Khalil et al., 2017; Wang et al., 2019; Layalle et al., 2021). Highly fragmented mitochondria have also been found among the axons of MNs in a D. melanogaster model expressing TDP-43 (Layalle et al., 2021). Sigma-1 receptor enhances the resistance of D. melanogaster to oxidative stress and exerts positive effects on motor activity and ATP levels (Couly et al., 2020).
In TDP-43 D. melanogaster models, significant changes were observed in glucose metabolism, including an increase in pyruvic acid levels. Among the MNs of TDP-43 D. melanogaster models, the mRNA level of phosphofructose kinase (an enzyme that controls the rate of fermentation) significantly increased (Manzo et al., 2019; Layalle et al., 2021). A high sugar diet can improve the exercise and life defect induced by TDP-43 protein lesions among MNs or/and glial cells, but it does not improve the muscles lesions in the TDP-43 D. melanogaster models, indicating that the metabolic disorders participate in the neural systems damage (Manzo et al., 2019). In addition, it has been revealed that some changes in the lipid metabolism, such as the reduction of both carnitine shuttle and lipid β oxidation, occur among TDP-43 D. melanogaster models (Layalle et al., 2021).
Among TDP-43 transgenic D. melanogaster models, other factors were found to have an impact on TDP-43. For example, the inhibition of protein tyrosine kinase 2 (PTK2) significantly reduces ubiquitin aggregation and cytotoxicity of TDP-43. Non-phosphorylated sequencesome 1 (SQSTM1) inhibits the accumulation and neurotoxicity of insoluble polyubiquitin proteins induced by TDP-43 overexpression in neuronal cells. TANK-binding kinase 1 (TBK1) participates in PTK2-mediated SQSTM1 phosphorylation. Therefore, the PTK2-TBK1-SQSTM1 axis plays a key role in the pathogenesis of TDP-43 by modulating neurotoxicity caused by damage to the ubiquitin-proteasome system in the TDP-43 transgenic D. melanogaster model (Lee et al., 2020). Recently, it was found that the generation of reversible droplet-like nuclear bodies (Wang et al., 2020), the overexpression of long non-coding RNA nuclear enriched abundant transcript 1–1 (Matsukawa et al., 2021), Mucuna pruriens and Withania somnifera (Maccioni et al., 2018; Paul et al., 2021; Zahra et al., 2022), and the production of CG5445 (a previously uncharacterized D. melanogaster gene) (Uechi et al., 2018, p. 3) alleviate the cytotoxicity of TDP-43 in neurons and improve movement or eye symptoms in the TDP-43 transgenic D. melanogaster model.
Gemin3 (DDX20 or DP103) is a dead-box RNA helicase that participates in a variety of cellular processes. The combination of Gemin3 and TDP-43 or FUS destruction aggravates vitality defects, motor dysfunction, and muscle atrophy while inhibiting the overgrowth of NMJ in the TDP-43 transgenic D. melanogaster model (Cacciottolo et al., 2019). A 3-fold knockdown of the lethal gene inhibitor (elongation factor of RNA polymerase II) significantly inhibited the morphological defects of the compound eye and medial retinae induced by TDP-43 in TDP-43 transgenic D. melanogaster models. D. melanogaster has five histone deacetylases (HDAC), among which Rpd3 knockdown significantly inhibits the rough eye phenotype caused by hTDP-43 in the TDP-43 transgenic D. melanogaster model (Yamaguchi et al., 2021). TBPH deficiency leads to larval death and reduced HDAC6 levels (Braems et al., 2021). Among the many single or double hydrophilic tag conjugating peptides D1-D8, D4 has the strongest ability to degrade TDP-43 protein and can penetrate the cell wall in short periods, inducing TDP-43 protein degradation in a dose- and time-dependent manner (Gao et al., 2019).
Reverse transposable element (RTE) expression was strongly upregulated in the head of TBPH-null D. melanogaster. TBPH regulates the silencing mechanism of small interfering RNA inhibiting RTE, and TBPH modulates Dicer 2 expression levels through direct protein mRNA interaction in TDP-43 transgenic D. melanogaster models (Romano et al., 2020). TBPH also interacted with VCP to inhibit VCP-induced degeneration in a TDP-43 transgenic D. melanogaster model (Braems et al., 2021).
4.3 D. melanogaster carrying FUS mutationsAlthough FUS mutations obviously affect MNs as well as other neurons found in FUS transgenic D. melanogaster models, its pathogenic mechanism of proteinopathy remains largely unknown. To study the alterations of FUS-associated functions in neurodegenerative diseases, including ALS, we reproduced the FUS transgenic D. melanogaster model expressing mutant human FUS (hmFUS), such as R518K, R521C, and R521H. These models cause a severe neurodegeneration in D. melanogaster eyes, whereas WT human FUS (hFUSWT) expression only led to mild neurodegeneration (Lanson et al., 2011).
Both movement defects and premature death have been observed in mutant FUS transgenic D. melanogaster. Moreover, mutant FUS overexpression increased the accumulation of the FUS protein. Causative roles on ALS-related hFUS mutants including both R524S and P525L in the FUS transgenic D. melanogaster model have also been described (Chen et al., 2011). Among the D. melanogaster models overexpressing WT and ALS-related FUS mutations among various neuron subsets, such as photoreceptors, mushroom bodies, and MNs, progressive age-related neuronal degeneration, such as axon loss, morphological alterations, and functional impairment of MNs, has been observed. The model of Cabeza (hFUS D. melanogaster homolog)-deficient D. melanogaster is used to study FUS functions, and the results showed both reduced lifespan and locomotor defects compared to controls. It was also found that transferring hFUSWT gene into this model could fully recover these phenotypes, but in this model co-expressing the ALS-related mutant FUS proteins, the phenotypes of reduced lifespan and locomotor defects were not recovered. This suggests that ALS-linked FUS mutations are toxic in FUS transgenic D. melanogaster models (Walker et al., 2011). In a transgenic D. melanogaster model coupled FUS with TDP-43 gene in neurons, it was demonstrated that FUS acts on ALS pathogen-associated effects together with TDP-43 via a common genetic pathway. Moreover, FUS and TDP-43 exert synergistic effects on RNA-dependent complexes. These models show that FUS and TDP-43 exert partial damage during the pathogenesis of ALS (Layalle et al., 2021).
D. melanogaster FUS homolog Cabeza is extensively expressed in the majority of tissues, including the neural system. Most Cabeza-D. melanogaster models with Cabeza functional loss are fatal, and only a few D. melanogaster are able to develop into adulthood, but exhibit severely shortened lifespans and motor disorders. Importantly, co-expression of wild-type Cabeza in the neurons of Cabeza-mutant D. melanogaster models can repair climbing and flight defects. Compared with Cabeza-mutant D. melanogaster models, larvae overexpressing Cabeza/FUSWT exhibited opposite NMJ electrophysiological phenotypes, characterized by reduced excitatory junction potentials and miniature excitatory junction potential amplitudes. In addition, it was found that the D. Melanogaster model may have a self-inhibitory mechanism for FUS/Cabeza, as the overexpression of FUS reduces the intracellular Cabeza content (Zhang et al., 2018a). D. melanogaster overexpressing hFUSWT exhibits abnormal eye morphology, shortened lifespan, eclosion defects, climbing defects, a reduced number of synaptic buttons, a reduced active region of the NMJ, and axonal degeneration (Azuma et al., 2018). hFUS is overexpressed in photoreceptor neurons in the eyes of D. melanogaster, resulting in mild retinal degeneration, rough eye surfaces, and a decrease in red pigment. The changes in the different FUS regions have different effects, such as in HEK293 cells, where the C-terminal truncated FUS chelates the FUSWT protein from the nucleus to the cytoplasm, thereby exacerbating FUS-induced retinal degeneration. The FUS-P525L mutation exacerbates FUS-induced retinal degeneration by increasing the FUS cytoplasmic distribution (Matsumoto et al., 2018; Layalle et al., 2021).
The arginine residue in the low-complexity domain of the FUS C-terminus is necessary for the maturation of FUS, while the N-terminal glutamine-glycine-serine-tyrosine (QGSY)-rich region (amino acids 1–164) and the C-terminal RGG2 domain are necessary for the toxicity of FUS, including the N-terminal QGSY, prion-like domains, and C-terminal RGG2 domains. It can distinguish between FUS and Cabeza toxicity based on its QGSY domain (Bogaert et al., 2018). The expression of different FUS mutations (R521C, 521H, 518K, R524S, or P525L) resulted in more severe rough-eye phenotypes in FUS transgenic D. Melanogaster models. FUS expression triggers Hippo activation and c-Jun N-terminal kinase signaling, leading to neuronal degeneration in FUS transgenic D. Melanogaster models. In addition, the Hippo signaling pathway is a modifier of Cabeza knockdown phenotypes (Layalle et al., 2021).
In FUS transgenic D. melanogaster models, it has been shown that inhibiting nuclear output can reduce hFUS-induced toxicity, thereby confirming the involvement of nuclear-cytoplasmic transport in the pathogenesis of ALS. It is worth noting that, recently, a series of complex enhancers and inhibitors have been found for ALS modifying factors screened in two different transgenic D. melanogaster models carrying human mutant FUS genes (Liguori et al., 2021). In the Cabeza knockdown D. melanogaster models, there are 14 inhibitory mutations knockdown models that effectively inhibit rough eye phenotypes caused by Cabeza knockdown. Mutations in Chameau and N-alpha-acetyltransferase 60 inhibit locomotion and cause morphological defects in NMJ synapses caused by Cabeza knockdown (Yamaguchi et al., 2021). FUS is similar to TDP-43 gene and regulates synaptic transmission among D. melanogaster NMJ; synaptic transmission defects precede the degeneration and loss of MNs in FUS transgenic D. melanogaster models. The nuclear-cytoplasmic transport proteins exportin-1 and nucleoporin-154 are modifiers of FUS toxicity, which prevent FUS-induced toxicity and reduce apoptosis of ventral nerve cord neurons (Braems et al., 2021).
In third-instar larvae of a Cabeza mutant D. melanogaster model, it was shown that the Cabeza mutant leads to an increase in induced and spontaneous neurotransmitter release in the NMJ (Zhang et al., 2018a). In addition, overexpression of hFUS led to a decrease in presynaptic activity areas and impaired synaptic transmission in hFUS transgenic D. melanogaster model. The loss and acquisition of FUS function in FUS and Cabeza transgenic D. melanogaster models may be achieved through dominantly negative mechanisms or the downregulation of endogenous Cabeza, which are mediated by interference with vesicular and mitochondrial transportation. This indicates that the Cabeza mutation leads to developmental and motor defects, which become more severe with age (Walters et al., 2019). Mutant FUS or D. melanogaster homolog Cabeza expressing type IV dendritic neurons leads to FUS or Cabeza cytoplasm mislocalization and axon transportation toward the pre-synapse end in FUS or Cabeza transgenic D. melanogaster models. Moreover, FUS or Cabeza overexpression leads to the progressive loss of neuronal projections and the reduction of synaptic mitochondria, while a large number of calcium transients appear in synapses by controlling the expression of calcium channels. In addition, mutant FUS overexpression results in a decrease in presynaptic synaptic-binding proteins, specifically disrupting axonal transport and inducing excessive excitability. Therefore, the mutation FUS/Cabeza damages the axon transportation of synapse vesicular proteins, thereby reducing the levels of synaptic-binding proteins, which are the main components of presynaptic release mechanisms. The overexpression of FUS can disrupt mitochondrial transport in D. melanogaster MNs, and an increase in FUS transport during neural biological processes alters the focal levels of synapse transcription or induces the formation of stress particles, thereby disrupting local translation and causing synaptic hyperexcitability (Machamer et al., 2018).
Karyopherin beta-2, also known as Kap-β-2 or transportin-1, participates in proline/tyrosine nuclear localization signal (PY-NLS), which inhibits and reverses the FUS fibrosis. In addition, it is worth noting that Kap-β2 can prevent RNA binding protein (RBP) with PY-NLS from accumulating among stress granules, restore the nucleus RBP localization and function, and rescue the neurodegeneration induced by ALS-related FUS in the FUS transgenic D. melanogaster models. The increase of Kap-β2 saves neurodegeneration and lifespan related to FUS in the FUS transgenic D. melanogaster models and reduces the accumulation of FUS in stress granules (Guo et al., 2018).
Knockdown of heat shock response gense in the D. melanogaster model expressing hFUSWT ω-LncRNA can reduce the level of hFUSWT mRNA and induce the generation of insoluble inclusion bodies comprising non-toxic hFUSWT proteins and lysosomal-associated membrane protein 1 in cytoplasm (Yamaguchi et al., 2021). Nuclear chromatin-binding protein (Xrp1), the main modifier of Cabeza mutative phenotypes, is strongly upregulated in the Cabeza-mutant D. melanogaster model, effectively reducing motor and life defects. Moreover, the Cabeza-mutant D. melanogaster model exhibits an imbalance in gene expression, and the Xrp1 heterozygosity can alleviate this (Mallik et al., 2018). Several genes, including TDP-43, Transportin-1, TER94/VCP, arginine methyltransferase, and the mitochondrial companion Hsp60 have been shown to interact with FUS/Cabeza genetically in the FUS/Cabeza transgenic D. melanogaster model (Zhang et al., 2018a).
4.4 D. melanogaster carrying C9orf72 mutationsC9orf72 is a common pathogenic gene involved in ALS. The present study showed that different cellular processes, including transcription, translation, nuclear-cytoplasmic transport, and protein degradation, are involved in the pathogenesis of C9orf72 ALS (Liguori et al., 2021). The D. melanogaster genome does not contain a direct homolog of human C9orf72, thus, an ALS-linked D. melanogaster model related to C9orf72 was developed by expressing the GGGGCC (G4C2) repeat (Yamaguchi et al., 2021). Although C9orf72 in ALS patients usually comprises over hundreds of G4C2 repeat sequences, the overexpression of 30–58 G4C2 repeat sequences in D. melanogaster eyes or MNs is sufficient to result in neuronal degeneration. Based on the expression level and time, the G4C2 repeat or arginine-rich dipeptide repeat (DPR) expressed among MNs leads to serious NMJ defects in the third-instar larvae of the C9orf72 G4C2 repeat transgenic D. melanogaster model (Zhang et al., 2018a). The amplification of the G4C2 hexanucleotide repeat expansion (HRE) of C9orf72 leads to the loss of C9orf72 function by inhibiting transcriptional extension and splicing of the first intron (Zhang et al., 2018a). The characteristic pathological hallmark observed among the different tissues of C9orf72 ALS, including MNs, is RNA lesions. RNA lesions are the result of HRE transcription, leading to the accumulation of repetitive RNA aggregates, usually located in the nucleus or cytosol (Layalle et al., 2021). This suggests that RNA-carrying HRE do not cause toxicity, but that the arginine-rich DPR sequence encoded by the HRE sequence mediates neurotoxicity (Moens et al., 2018). Consistent with this, the abnormal translation of HRE leads to the accumulation of DPR protein in the brains of ALS patients (Azoulay-Ginsburg et al., 2021). When comparing the effects of RNA expression among the HRE transgenic D. melanogaster model, only DPR proteins containing arginine have neurotoxicity on the G4C2 motifs encoding different dipeptide combinations (Bolus et al., 2020).
Poly (glycine-arginine) (GR) is a common genetic cause of ALS and FTD, occupying forty percent of fALS cases (Cunningham et al., 2020). Partial functional loss of an essential DNA repair protein Ku80 inhibits retinal degeneration induced by poly (GR) in poly (GR) transgenic D. melanogaster models. In the neurons of D. melanogaster model expressing poly (GR) and in patients with C9orf72, the Ku80 expression is significantly increased. Elevated Ku80 expression contributes to GR aggregation and induces neurodegeneration in a poly (GR) transgenic D. melanogaster model (Lopez-Gonzalez et al., 2019).
Arginine-rich DPR is a highly toxic product derived from the C9orf72 repeat amplification mutation and is a common cause of fALS. However, their role in synaptic regulation and excitatory toxicity remains unclear. Applying C9orf72 DPRs with different toxic intensities revealed that arginine-rich DPRs induced selective degeneration of glutamate neurons in the C9orf72-DPR transgenic D. melanogaster model. Among them, the C9orf72-DPR can stimulate synaptic overgrowth and promote glutamate release. Furthermore, an increase in glutamate release from excitatory neurons expressing DPR not only causes excitotoxicity in postsynaptic neurons, including MNs, but also produces cell-autonomous excitotoxicity in presynaptic neurons (Xu and Xu, 2018).
Up-frameshift 1 (UPF1) is an RBP with helicase activity that reduces DPR-mediated toxicity. Overexpression of UPF1 significantly reduces the severity of known neurodegenerative phenotypes and poly (GR) content without changing the amount of repeat RNA, indicating that UPF1 has a neuroprotective effect on C9orf72-ALS (Zaepfel et al., 2021). The present study shows that UPF1 expression regulates the DPR- or HRE-induced neurodegenerative phenotypes of C9orf72-ALS transgenic D. melanogaster models, including eye symptoms (Xu et al., 2019; Ortega et al., 2020; Sun et al., 2020). In addition, DPR can directly bind to nucleoporins, and knockdown of karyopherin alpha3 in the C9orf72-ALS transgenic D. melanogaster model can enhance the toxicity of DPR (Walters et al., 2019; Braems et al., 2021). Furthermore, the loss of C9orf72 function disrupts autophagy and lysosomal functions in many types of cells, including MNs (Ji et al., 2017; Shi et al., 2018; Zhu et al., 2020).
The key regulatory factor of autophagy, which is refractory to Sigma P/sequestosome 1 [Ref(2)P/p62], is an effective inhibitor of ALS induced by G4C2 HRE in C9orf72 (Cunningham et al., 2020). The microphthalmia/transcription factor E family of transcription factors is a key regulator of autophagy-lysosome function. Transcription factor EB (TFEB) is a candidate therapeutic target for ALS (Cortes and La Spada, 2019). It was found that C9orf72-HRE impaired the microphthalmia-associated transcription factor/transcription factor EB nuclear input, disrupted autophagy, and exacerbated the protein balance defect in the C9orf72 ALS transgenic D. melanogaster model. Increased TFEB activity may prevent neuronal death and promote recovery of neuronal function through different mechanisms in the C9orf72-ALS transgenic D. melanogaster model (Torra et al., 2018).
The G4C2 amplification repeat sequence and tau protein are traditionally considered to be related to partial clinical manifestations in several neurodegenerative diseases, such as ALS. Co-expression of the G4C2 repeat sequence and Tau can lead to the synergistic deterioration of rough eyes, movement functions, longevity, and abnormal NMJ morphology in the G4C2 repeat sequence and Tau co-expression transgenic D. melanogaster model. In addition, 30 G4C2 repeats increase tau phosphorylation levels (He et al., 2019). Moreover, the downregulation of D. melanogaster tau homologous protein can reduce neurodegeneration and motor lesions and prolong the lifespan of C9ORF72 G4C2 repeatedly expressing D. melanogaster model (Braems et al., 2021).
5 C. elegans modelsThe C. elegans genome was the first multicellular organism to be completely sequenced in 1998 (C. elegans Sequencing Consortium, 1998). C. elegans contains ~20,000 genes that are highly similar to the 25,000 human genes. Therefore, it is considered as an ideal model for analyzing the genetic and molecular mechanisms underlying neuronal development, function, and disease. Moreover, more than 42% of human disease-linked genes have an organology with C. elegans, suggesting that almost all biochemical pathways have been conserved throughout the evolution of C. elegans and humans (Culetto and Sattelle, 2000). C. elegans was the first model used to study the biochemical pathways of human diseases.
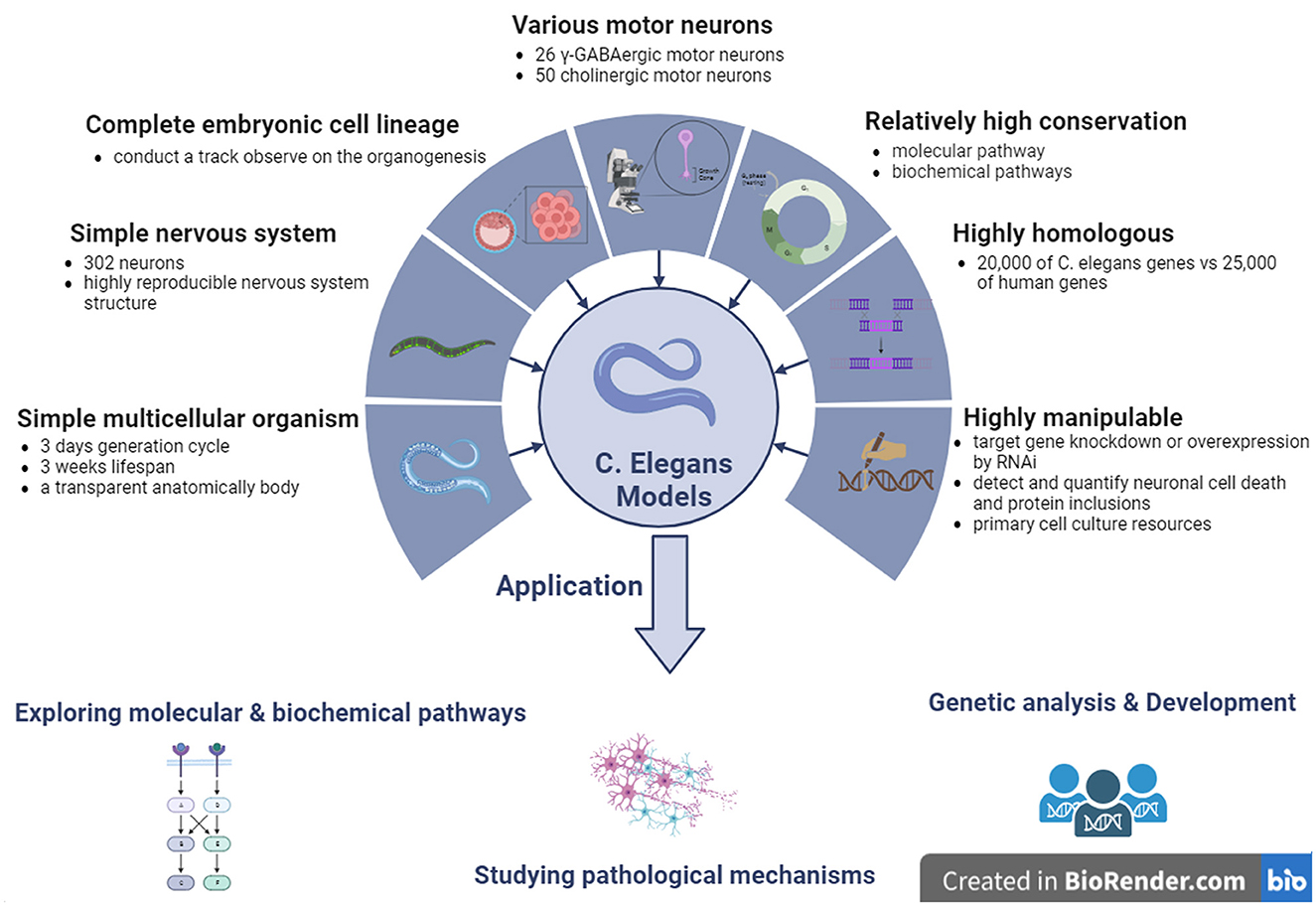
C. elegans is genetically tractable, which indicates that its genome can be easily manipulated. This characteristic is particularly advantageous in ALS research, in which the identification of genetic factors contributing to the disease is essential. Researchers can introduce specific mutations associated with ALS into the C. elegans genome to observe the resulting phenotypic changes. This approach helps elucidate the genetic basis of ALS and identify potential therapeutic targets (Clarke et al., 2018). Twenty-six neurons in five different neuronal types are gamma-aminobutyric acid (GABA)-ergic neurons in C. elegans (McIntire et al., 1993). Among them, 19 GABAergic neurons, called type D neurons, exert regular locomotion by providing dorsal-ventral cross inhibition to the body wall muscle (White et al., 1986; McIntire et al., 1993). In addition, C. elegans contains more than 50 cholinergic MNs that control locomotion (Liu et al., 2020). Therefore, C. elegans transgenic models are useful for studying the pathological alterations related to locomotor dysfunction. C. elegans is a hermaphrodite, ~1 mm in length, with almost 3 days of short generation cycles, and ~300 progenies in a large brood. It has a short lifespan of ~3 weeks and undergoes rapid generation cycles, allowing for quick observation of the effects of genetic manipulations across multiple generations. In ALS studies, this characteristic enables researchers to assess the progression of neurodegeneration and the inheritance patterns of genetic mutations associated with the disease in a relatively short timeframe (von Mikecz, 2022). C. elegans has a transparent anatomical body that can be used to visualize all cell types at developmental stages (Brenner, 1974). The transparent body of C. elegans is another advantage, especially for the visualization of cellular and subcellular events. Researchers can observe the development and degeneration of neurons in real-time, providing valuable insights into the mechanisms of ALS pathogenesis. This transparency aids in monitoring changes, such as protein aggregation, cellular death, and alterations in neuronal morphology. The experimental techniques available for C. elegans make it easy to observe and quantify cellular events such as neuronal cell death and protein inclusion. This is critical for ALS research, in which an understanding of the dynamics of these events is essential. The C. elegans transparent body, combined with advanced imaging techniques, facilitates detailed analysis of cellular changes associated with ALS. Another advantage of the C. elegans model is the ease of observation and quantification of neuronal cell death and protein inclusion using experimental techniques (Ma et al., 2022).
The whole embryo cellular lineages of C. elegans are completely clear, which makes it possible to track organogenesis from the earliest stage of embryo formation to the terminal stage of organ differentiation and morphogenesis (Sulston et al., 1983). In addition, the nervous system of C. elegans is very simple; it possess 302 neurons among 959 cells in an adult C. elegans body. Each neuron is located in a unique anatomical position (Sulston et al., 1983). Twenty neurons are located in the larynx, while the 282 remaining neurons are located in various ganglia, from head to tail, along the main longitudinal axon tract in the ventral spinal cord. Among the neurons that develop during embryogenesis, 80 MNs develop at the post-embryonic stage. C. elegans has a relatively simple nervous system with 302 neurons, making it an excellent model for studying neural circuits and functions (Takeishi et al., 2020). Understanding the molecular and cellular events that underlie neurodegeneration is crucial for ALS research. The simplicity of the C. elegans nervous system allows researchers to map and monitor individual neurons, thereby facilitating the identification of key genes and pathways involved in ALS pathology (Gois et al., 2020).
The structures of the C. elegans nervous system were highly reproducible in each generation, as described previously, and high-resolution images were obtained by electron microscopic reconstruction. Approximately 5,000 chemical synapses, 2,000 neuromuscular junctions, and 500 gap junctions have been described, and all connections of the entire neuronal circuit have been mapped in C. elegans models (White et al., 1986). In C. elegans models, it is relatively easy to achieve target gene knockdown or overexpression by RNAi by injecting dsRNA into the unique genes of interest, simply soaking C. elegans in the dsRNA medium or feeding C. elegans the bacterium carrying the targeted dsRNA (Fire et al., 1998; Maeda et al., 2001). In addition, primary culture resources for neurons and muscle cells can also be obtained by dissecting C. elegans, which can further optimize and stabilize the growth of embryonic cells (Christensen et al., 2002).
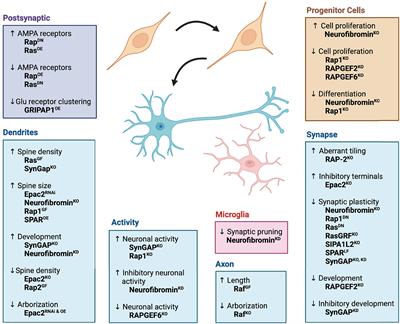
Although many disease-relevant genes have been identified in neurodegenerative disorders, including ALS, the mechanisms underlying the dysfunction and death of selective MNs remain poorly understood. Based on this, molecular conservation in the neuronal signaling pathway between invertebrates and vertebrates is high, and C. elegans contains almost all known signaling and neurotransmitter systems found in the nervous system of mammals (Bargmann, 1998). Therefore, C. elegans models have generally been used by researchers to investigate the mechanisms underlying neurodegenerative diseases, including ALS. Previous studies on C. elegans have provided valuable data and evidence for elucidating the molecular pathways involved in many human diseases, including ALS. Because of these advantages, C. elegans has attracted many researchers to use it as an in vivo model for researching pathological mechanisms in neurodegenerative diseases such as ALS, providing some perspective evidence for identifying potential pathogenic targets and developing new therapeutic measures against human diseases such as ALS (Figure 4).
Figure 4. The features and applications of C. elegans genetic models in ALS. ALS, amyotrophic lateral sclerosis; C. elegans, Caenorhabditis elegans; γ-GABA, gamma-aminobutyric acid.
5.1 C. elegans carrying SOD1 mutationsSince C. elegans ALS models were first established in 2001 (Oeda et al., 2001), a series of C. elegans models have been developed, and many potential mechanisms associated with ALS pathogenesis have been identified. Among them, the hmSOD1 C. elegans model was produced by transferring wild-type human SOD1 (WThSOD1) and familial ALS (fALS) SOD1 mutations, such as A4V, G37R, and G93A, by controlling several promoters of the heat shock protein (HSP)-16.2 and myosin heavy chain 3 (myo-3) genes. The hmSOD1 transgenic C. elegans model controlled by the inducible HSP-16.2 promoter expresses hmSOD1 in nearly all tissues, including neurons, and the hmSOD1 C. elegan models controlled by the muscle-specific promoter of myo-3 largely express hmSOD1 among all muscles besides the pharynx.
The universal expression of hmSOD1 in C. elegans blocks natural biological responses to oxidative stress and induces the aggregation of toxic proteins, while the expression of hmSOD1 in all C. elegans neural systems leads to motor defects and neuron transmission damage in C. elegans models. The establishment of a SOD1 single copy (A4V, H71Y, L84V, G93A, and G85R) mutation knockout C. elegans model can analyze the unique effect of the special mutation on the degeneration of cholinergic and glutamic MNs in C. elegans models. These findings indicate that the pathogenesis of ALS is a neuronal subtype-specific gain of toxic functions, as well as a loss of physiological functions (Liguori et al., 2021). Moreover, loss of SOD1 function is the main cause of glutamate neuron degeneration after oxidative stress in C. elegans models, and the toxic functions gaining of SOD1 protein may cause cholinergic MNs degeneration in C. elegans models (Baskoylu et al., 2018). In the ALS-like hmSOD1 C. elegans model containing the ALS G85R mutation, a relatively high level of hmSOD1 was expressed in the neurons of C. elegans, resulting in movement defects of C. elegans expressing SOD1 G85R (Baskoylu et al., 2018).
Although morphological abnormalities of neurons and discernible survival or behavioral changes were not observed in the C. elegans model of hmSOD1 expression, the hmSOD1 C. elegans model demonstrated decreased resistance to paraquat-induced oxidative stress, significantly reducing the degradation ability of hmSOD1 proteins, leading to the abnormal accumulation of hmSOD1 proteins among muscular cells. Moreover, the final pathological phenotypes of hmSOD1 proteins in the C. elegans models were similar to the pathologically altered features observed in the post-mortem tissues of ALS patients.
The transgenic C. elegans model of entire neuronal expression of hSOD1 G85R using the synaptobrevin (snb-1) gene promoter coupled with yellow fluorescent protein (YFP) exhibits serious movement defects and paralysis (Wang et al., 2019). The observed phenotypes in this C. elegans model corr
留言 (0)