
記住我
The genus Dendroctonus (Curculionidae: Scolytinae) is a Holarctic taxon composed of 21 nominal species, 19 of which are found in the Nearctic region from Alaska to Honduras and two in the Palearctic region in Europe and Asia (Victor and Zuniga, 2016; Valerio-Mendoza et al., 2019), although recently a North American taxon was indirectly introduced into China (Yan et al., 2005). Some of the species of these bark beetles are well-known disturbance agents of forest ecosystems, as their populations frequently experience outbreaks due to varying exogenous factors including droughts, warming climate, attributes of forest stands, topography, soil type, and humidity, resulting in the mortality of thousands to millions of trees with drastic effects on ecosystem structure, function, and composition (Kurz et al., 2008; Raffa et al., 2015).
The life cycle of Dendroctonus beetles consists of three phases: a short dispersal period, host tree colonization, and subcortical growth and development by new offspring. This last phase takes place mainly under the tree bark where colonizing adults and larvae feed on phloem tissue of the genera Larix, Pseudotsuga, Picea, and especially Pinus (Six and Bracewell, 2015). In the subcortical environment, Dendroctonus species are associated with many phoretic ectosymbiotic (e.g., filamentous fungi, yeasts and bacteria, nematodes, and mites) and endosymbiotic communities (e.g., yeasts and bacteria) (Hofstetter et al., 2015). These symbiotes are carried by beetles to trees on the external surface of their bodies, in specialized morphological structures, such as mycetangia and nematangia, and within the digestive system, including mouthparts and the alimentary canal (Beaver, 1989). Despite the strong association of Dendroctonus spp. with microbial communities, there is no evidence that they are acquired by vertical transmission, suggesting that they are acquired de novo in each new generation from the subcortical environment (Six and Bracewell, 2015; Hernández-García et al., 2017; Gonzalez-Escobedo et al., 2018; Vazquez-Ortiz et al., 2022).
Interactions between Dendroctonus species with their symbiotes have been studied intensively for many years (Klepzig and Six, 2004; Davis, 2015), even though these interactions are complex and dynamic in space and time (Six and Klepzig, 2021). The general pattern that emerges from earlier studies is that microbial associations are involved in several ecological processes that may enhance beetle fitness during the process of tree colonization and brood development. However, the interaction with specific members of microbial communities and their ecological role in many cases is not known. For example, some Dendroctonus are associated with both teleomorph and anamorph forms from several Ascomycetes (e.g., Ophiostoma/Sporothrix, Ceratocystis, Ceratocystiopsis, Grosmannia/Leptographium) and Basidiomycetes (e.g., Entomocorticium) (Paine et al., 1997; Harrington, 2005), which can affect the beetle lifecycle vis-à-vis obstruction of phloem and xylem tracheids, toxic terpenes detoxification, the regulation of insect development, the inhibition of pathogenic microorganisms, and nutritional supplementation (Bentz and Six, 2006; Davis et al., 2011; DiGuistini et al., 2011; Dai et al., 2022).
Bacteria are also common microbial associates of Dendroctonus spp., and in vitro studies show that the members of the genera Arthrobacter, Bacillus, Brevundimonas, Cellulosimicrobium, Cellulomonas, Janibacter, Kocuria, Leifsonia, Methylobacterium, Paenibacillus, Ponticoccus, Pseudomonas, Pseudoxanthomonas, Rahnella, Serratia, Stenotrophomonas, and Sphingomonas have abilities that match or complement many of the same ecological functions as fungi (Scott et al., 2008; Adams et al., 2009; Morales-Jiménez et al., 2009, 2012, 2013; Boone et al., 2013; Hu et al., 2014; Mason et al., 2014; Cano-Ramírez et al., 2016; Xu et al., 2016; Briones-Roblero et al., 2017a; Pineda-Mendoza et al., 2022).
Unlike filamentous fungi and bacteria, the ecological role of yeasts in the bark beetle is less well understood, although yeasts were the first group of microbes to be studied systematically in bark beetles (Davis, 2015). Several commonly isolated yeast species, including Kuraishia capsulata, Ogataea pini, Nakazawaea holstii (= Pichia holstii), Wickerhamomyces bovis (= Pichia bovis), W. canadensis, Candida oregonensis, Cyberlindnera americana, and Zygoascus sp., have abilities to degrade terpenes, starch, and lipids (Hunt and Borden, 1990; Briones-Roblero et al., 2017b), produce semiochemicals (Davis et al., 2023), or inhibit the growth of filamentous fungi (Davis et al., 2011). Many other yeasts have been isolated from mycetangia, gut, frass, and galleries of Dendroctonus, but their ecological functions have not been tested (Rivera et al., 2009; Lou et al., 2014; Dohet et al., 2016).
The above studies performed with cultivation-dependent methods demonstrated the functional capacities of specific members of assemblages, but they did not allow for an estimate of overall fungal biodiversity or phylogenetic diversity in beetle-microbe assemblages and, as a result, underestimated the diversity. They also did not elucidate the integral functional capacity of assemblages, or the importance of specific partners in them, as well as their relationships among one another. However, the advent of next-generation sequencing technologies has enabled more comprehensive and detailed analysis of Dendroctonus microbial communities, including the exclusiveness and persistence of microbes into communities, the analysis of the community structure across the life cycle of whole insects, geographic sites, beetle species, body parts, and field insects vs. laboratory rearing insects, as well as the identification of genes involved in terpene degradation (Adams et al., 2013; Durand et al., 2015, 2019; Dohet et al., 2016; Briones-Roblero et al., 2017b; Hernández-García et al., 2017, 2018).
Thus far, little is known about the gut mycobiome of Dendroctonus bark beetles (Rivera et al., 2009). Some studies have used culture-independent techniques to describe the fungal community composition and changes across the life cycle of whole insects (Lou et al., 2014; Dohet et al., 2016; Durand et al., 2019), with only one earlier study analyzing the composition of fungal assemblages in the mycetangia of Dendroctonus frontalis species complex (Vazquez-Ortiz et al., 2022). In this study, we aim to characterize and compare the gut-associated fungal community of 14 species of the genus Dendroctonus through the analysis of the internal transcribed spacer 2 (ITS2) region. Specifically, we describe how the community structure of the gut-associated fungal assemblages differs within and among species (α- and β-diversity) and examine whether a common mycobiome is shared or not among beetle species. Finally, we predict the functional ecological role of fungal assemblages and characterize interspecific linkages among fungi associates.
2 Materials and methods 2.1 Insect collection, dissection, and DNA extractionDuring the summer of 2019, emerging adult insects of 14 species of Dendroctonus were collected directly from the galleries of infested pine and spruce trees in different localities from North and Central America (Table 1). Live insects were taken from galleries with sterile forceps and stored at 4°C in sterile polycarbonate Magenta™ vessels GA-7 (Sigma-Aldrich, United States) for transport to the laboratory. Insects collected in the United States were shipped inside sterile vials in absolute ethanol. All samples were processed immediately upon arrival at the laboratory. Taxonomic identification was verified according to the work of Armendáriz-Toledano and Zúñiga (2017).
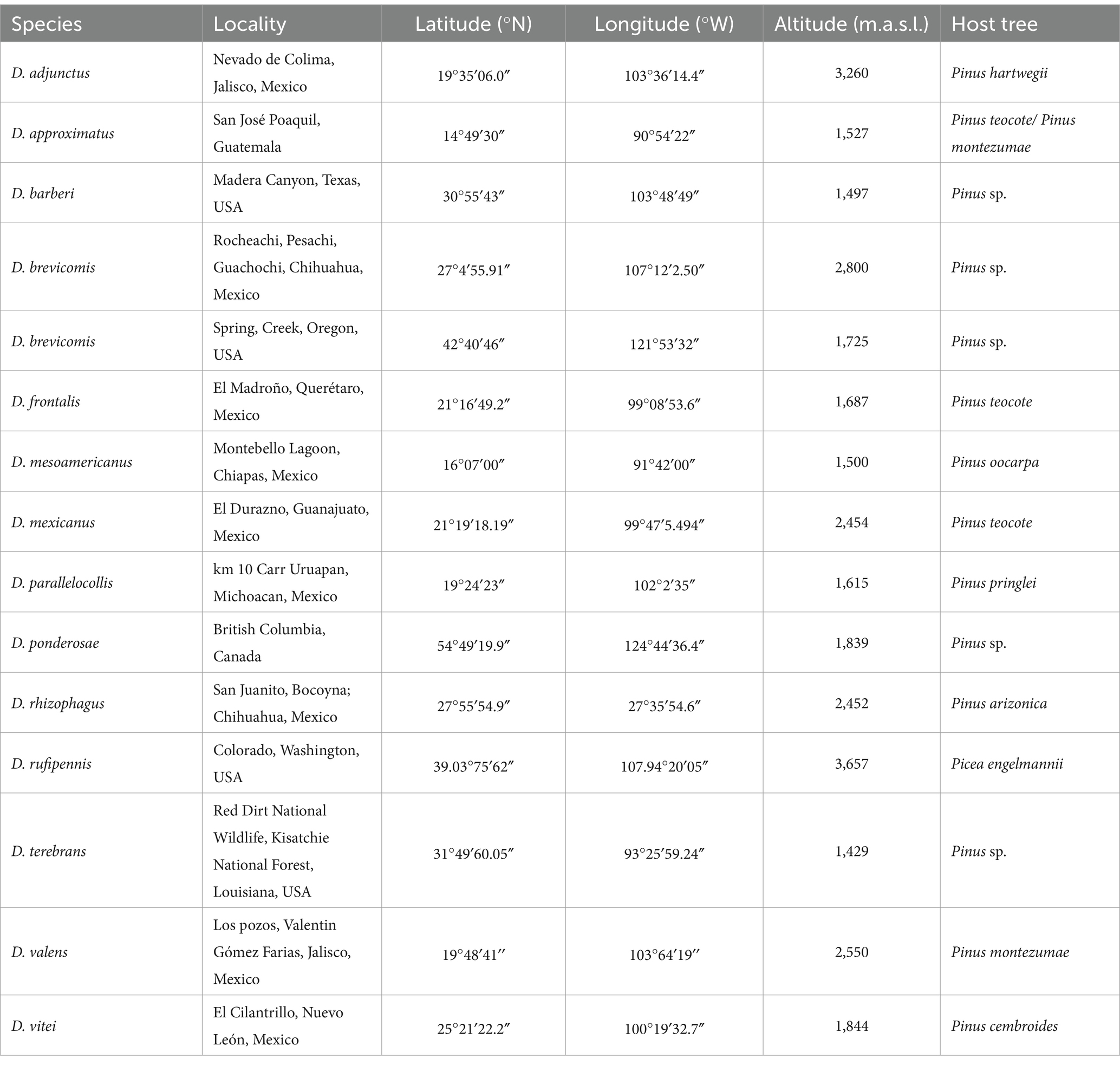
Table 1. Collection localities of the 14 species of the Dendroctonus genus sampled in the present study.
All insects were surface disinfected by immersion in the following solutions at 1 min intervals: detergent solution (10 mmol L−1 Tris–HCl pH 8.0, 1 mmol L−1 EDTA, 10 mmol L−1 NaCl, 1% SDS, and 2% Triton X-100), 70% ethanol solution, and then washed three times with sterile distilled water. Afterward, they were placed in sterile phosphate buffer (PBS, pH 7.4; 137 mmol L−1 NaCl, 2.7 mmol L−1 KCl, 10 mmol L−1 NaHPO4, and 2 mmol L−1 KH2PO4) for dissection under aseptic conditions using a stereo microscope (MZ6 Leica, Germany). A longitudinal incision was performed with forceps scissors and sterilized fine-tipped to extract the gut, removing the elytra, wings, and tergites (Briones-Roblero et al., 2017b). We did not consider the sex of insects because previous studies performed with bacteria and yeasts through culture-independent and culture-dependent techniques did not show significant differences in sexes (Rivera et al., 2009; Briones-Roblero et al., 2017a). Three biological replicates per species were independently carried out, each replicate integrated by a pool of 10 guts. Homogenization of samples was performed in 1.5 mL Eppendorf tubes with 500 μL of sterile PBS and sterile plastic pestles. The metagenomic DNA was obtained with the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s specifications, and their concentration and purity were evaluated in a NanoDrop™ 2000c Spectrophotometer (Thermo Scientific, Wilmington DE, USA).
2.2 Sequencing and data analysisThe ITS2 amplification was carried out with the universal primer pairs ITS3 (5′-GCA TCG ATG AAG AAC GCA GC-3′) and ITS4 (5′-TCC TCC GCT TAT TGA TAT GC-3′). We used this region because it is less variable in size than ITS1, but sufficiently variable in nucleotide content to discriminate ASVs (Taylor et al., 2016). The amplicon libraries were sequenced using a paired-end 2 × 300 bp Illumina MiSeq sequencer according to the protocols of Macrogen Inc. (Seoul, Korea). The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (Bioproject number PRJNA1074716). Raw paired-end reads were processed in Quantitative Insights Into Microbial Ecology (QIIME2) v.2023.2, last accessed June 2023 (Bolyen et al., 2019). To remove low quality and/or uninformative features, the raw read sequences were subjected to a denoising process consisting of quality filtering (Phred 30 score), trimming, dereplication, merging, and the removal of chimera sequences using the DADA2 plugin (Callahan et al., 2016). To determine amplicon sequence variants (henceforth called ASVs) at a 97% identity threshold, we used the pre-trained Naive Bayes classifier against the UNITE database (Kõljalg et al., 2020). To confirm ASV taxonomic identity, they were manually verified in GenBank. ASV contaminants from insects, mites, nematodes, and plants were removed from the data set. ASVs were aligned with the Multiple Alignment using Fast Fourier Transform program (MAFFT, Katoh and Standley, 2013) to infer an unrooted maximum-likelihood phylogenetic tree with IQ-TREE (Nguyen et al., 2015) using as the best model of nucleotide evolution TIM2f + I + G4 sensu Akaike criterion (-InL = 22909.196, I = 0.12, and G = 0.925). Given the size of the libraries was uneven and the observed abundance data were compositional, the ASV abundances were standardized using the centered log-ratio (CLR) transformation in CoDaPack v.2.02.21 (Comas-Cufí and Thió-Henestrosa, 2011). As fungi taxonomy is dynamic and constantly changing, in this study, we employ the current taxonomic names and the lineage or clade name following those given in the NCBI database.
2.3 Diversity analysis of fungal assemblagesThe sampling coverage was calculated using rarefaction curves and the Good’s coverage in QIIME2. ASV abundance at the phylum, class, order, family, and genus levels at 97% similarity was represented as bar plots using ggplot2 in the R v.4.2.1 package. The α-diversity of each fungal assemblage of Dendroctonus species was estimated with metrics Chao1 (species richness), Shannon-Weaver (diversity), Simpson and Simpson reciprocal (dominance) in QIIME2. To test for differences in α-diversity metrics, we used the Kruskal–Wallis test in QIIME2. Based on the presence or absence of fungal genera in all libraries, we inferred using UpSetR package v 1.4.0 (Conway et al., 2017) the total number of fungi, shared and unique, present in the gut of each beetle species, as well as a common mycobiome core (CMC) among them.
To infer the ability of some members representing the genera that were part of CMC, we downloaded genomes from NCBI and searched for genes involved in the degradation of fungal cell wall (chitin, glucan, and mannan) and plant cell wall (cellulose, hemicellulose, pectin, and lignin), as well as in the biosynthesis of essential amino acids. Gene prediction was carried out with AUGUSTUS v.2.5.5 (Stanke et al., 2004), and their functional annotation and metabolic pathways in which they participate delineated in the Kyoto Encyclopedia of Genes and Genomes (KEGG) database. In addition, the guild, the trophic mode, and the growth morphology of fungal ASVs in the gut of the Dendroctonus species were inferred using the FUNGuildR package (Nguyen et al., 2016), supplemented with information from the specialized literature.
β-diversity of gut fungal assemblages from Dendroctonus species was visualized using a principal component analysis (PCA) in scatterplot3d v.0.3–44 in the R package based on Aitchison distance (Aitchison, 1982; Aitchison et al., 2000), and weighted and unweighted UniFrac distances (Lozupone and Knight, 2005) using the CLR matrix in the phyloseq R package v.1.42.0 (McMurdie and Holmes, 2013). Significant differences in microbiome composition (β-diversity) between Dendroctonus species were performed by permutational multivariate analysis of variance (PERMANOVA) based on 9,999 random permutations in PAST v.4.10 (Hammer and Harper, 2001). In addition, to assess whether the composition of mycobiomes among within Dendroctonus species is similar, we performed the PERMDISP test with 10,000 permutations in QIIME2.
2.4 Co-occurrence analysis and nutritional and functional guildsTo determine the interactions among ASVs with ≥23 reads in each of the fungal assemblages of Dendroctonus species, a co-occurrence empirical network based on the random matrix theory was constructed using pairwise Spearman correlation (p < 0.05) in the molecular ecological network analysis (MENA) platform with default settings (Deng et al., 2012). To test whether the linkage among ASV interactions was due to non-random effects, a set of 100 random networks was generated from the empirical community structure. Significant differences between the empirical and the null random networks were evaluated using the Z test (p < 0.05). The random network was constructed with the Maslov-Sneppen method in MENA, which kept the number of nodes (taxa) and edges (connections) unchanged but rewired the positions of all links in the network. Network properties including modularity degree (M, a measure of the internal organization of the network into modules), average connectivity (avgK, average number of node connections within the network), average path distance (GD, average measure of the shortest paths between two nodes), and average clustering coefficient (avgCC, average measure of the extent to which nodes are grouped together in a network) were calculated in the MENA pipeline. The global network was visualized in the Cytoscape v.3.10.0 (Shannon et al., 2003). In addition, to understand the relative contribution of fungal genera to the structure and stability of the assemblages, we carried out a bipartite ecological analysis using the bipartite R package (Dormann et al., 2008). Two bipartite networks were built, the first with the most frequently occurring fungal genera and the second with the less abundant genera.
3 Results 3.1 Sequencing dataA total of 5,592,374 reads were obtained from 42 libraries from 14 Dendroctonus species. After quality control, 615,542 sequences were recovered, yielding a total of 248 fungal ASVs. Rarefaction curves and Good’s coverage (>99%) indicated that sampling effort for all libraries was appropriated to representatively capture taxonomic diversity (Supplementary Figure 1).
The results showed a total of four phyla, 16 classes, 34 orders, 54 families, and 71 genera with different relative abundances among Dendroctonus species. The phylum most abundant was Ascomycota (99.85%), followed by Basidiomycota (0.13%), Mucoromycota (0.006%), and Mortierellomycota (0.002%). Specifically, the relative abundance of yeasts and fungi at the family and genus level varied among insect species (Figures 1A,B). Among the families that stand out as the most abundant are Debaryomycetaceae (49.7%), Pichiaceae (19.5%), Phaffomycetaceae (17.53%), Aspergillaceae (9.5%), and Cladosporiaceae (1.07%); the remaining families represented 2.7% of the relative abundance, but all of them had relative frequencies of <1% (Figure 1A).
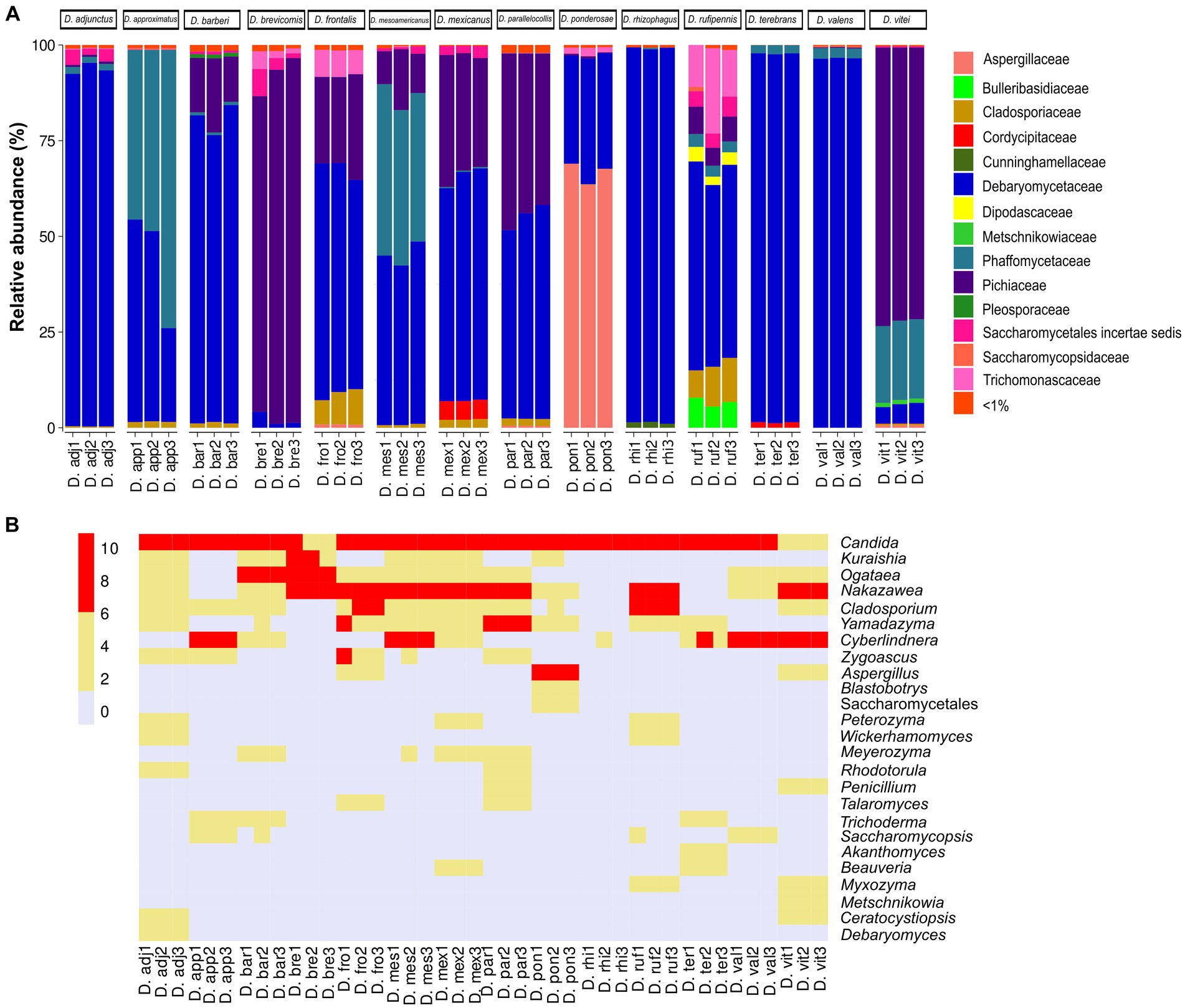
Figure 1. Fungal diversity and relative abundance of the gut assemblages associated with 14 species of the genus Dendroctonus. (A) A bar plot at the family level. (B) The heat map of the genera identified by bark beetle species. The color gradient shows the relative ASV’s abundance for each library.
In total, 32 genera of yeasts and 39 filamentous fungi were recorded, with the first being the more abundant than the second (Figure 1B), highlighting Candida (48.16%) as the most abundant in all Dendroctonus species, followed by Nakazawaea (18.13%), Cyberlindnera (17.44%), Aspergillus (9.44%), Ogataea (1.46%), Yamadazyma (1.42%), Cladosporium (1.07%), and other genera (2.88%) with <1% each of them of the total reads.
3.2 The analysis of fungal assemblage diversityAccording to Kruskal–Wallis and Dunn tests, the gut-associated fungal assemblages of Dendroctonus bark beetles showed statistically significant differences in species richness, species diversity, and dominance (p < 0.05; Figure 2). The higher species richness was found in assemblages of D. ponderosae, D. approximatus, and D. vitei, and the lowest species richness was found in those of D. valens, D. brevicomis, and D. rhizophagus. On the other hand, the gut fungal diversity in D. rufipennis, D. frontalis, and D. mesoamericanus assemblages was the most diverse with both metrics, whereas those from D. rhizophagus, D. terebrans, and D. valens were the least diverse. The number of dominant fungi varied among assemblages of bark beetle species from 2 to 5, with D. valens exhibiting the fewest dominant fungi in assemblages and D. rufipennis showing the most.
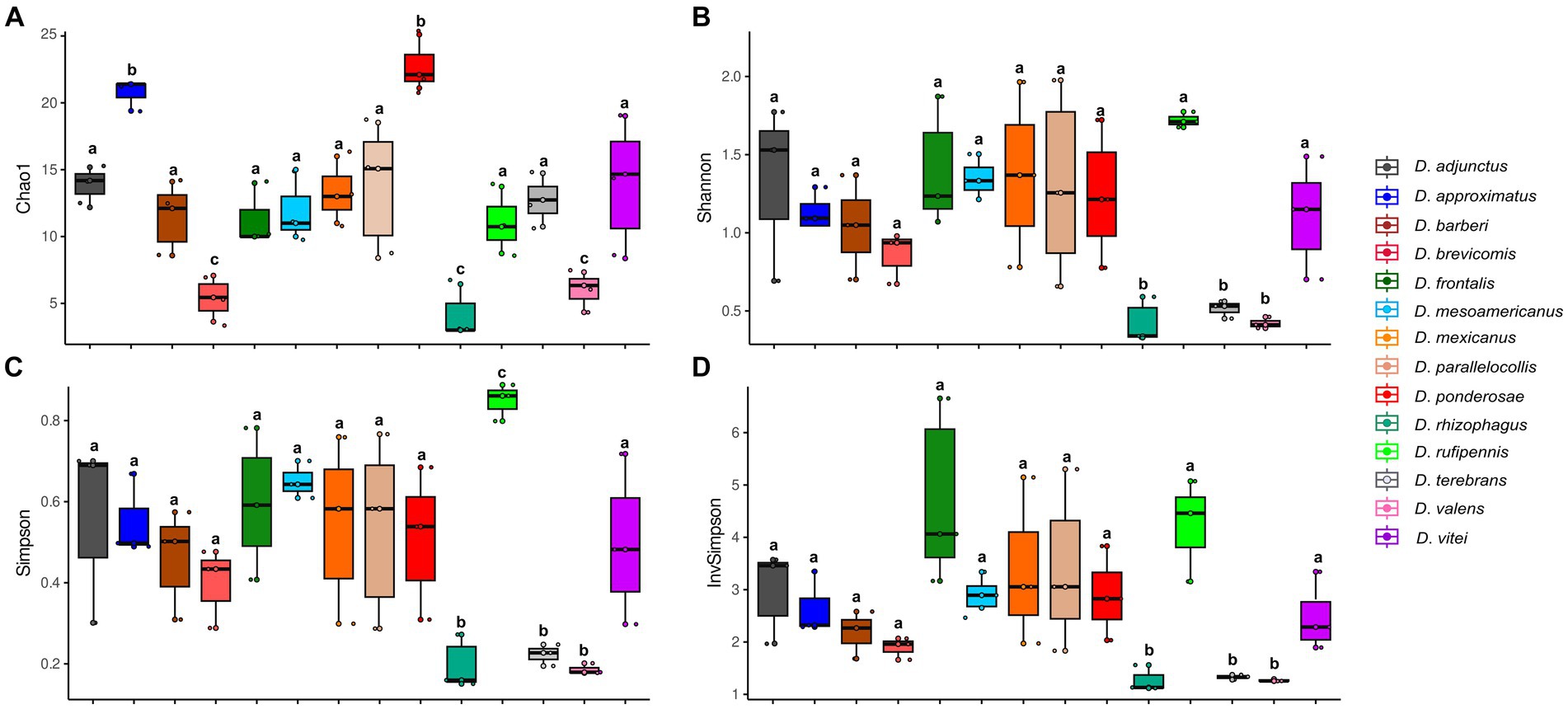
Figure 2. Alpha diversity indexes representing the fungal assemblage richness and diversity from 14 species of Dendroctonus genus. (A) Species richness by Chao1, (B) Shannon-Weaver diversity, (C) Simpson dominance, and (D) Simpson’s reciprocal diversity. Box plots not connected by the same letter are statistically different (p < 0.05).
Statistically significant differences in the composition of the gut fungal assemblages (β-diversity) were found among bark beetle species according to the PERMANOVA tests using Aitchison distance (F = 51.704; p = 0.001) or unweighted (F = 45.072; p = 0.001) and weighted (F = 116.36; p = 0.001) UniFrac distances. The first three components of the PCA based on Aitchison distance explained 26.8, 15.9%, and 11.6% of the total variations observed among the bark beetle samples, respectively. The distribution of bark beetle assemblages in this multidimensional space showed a disjunct pattern among species, but no clear differentiation among within-species replicates (Figure 3). This pattern was also observed in the PCA using unweighted and weighted UniFrac distances but with different percentages of variation explained by the first three principal components (unweighted, C1 = 44.4%, C2 = 15.2%, and C3 = 10.1%; weighted, C1 = 70.1%, C2 = 13.2%, and C3 = 5.9%) (Plots shown in Supplementary Figures 2A,B). The PERMDISP test showed that dispersions around the mean or centroid were homogenous across samples (F = 7.72; p = 0.076).
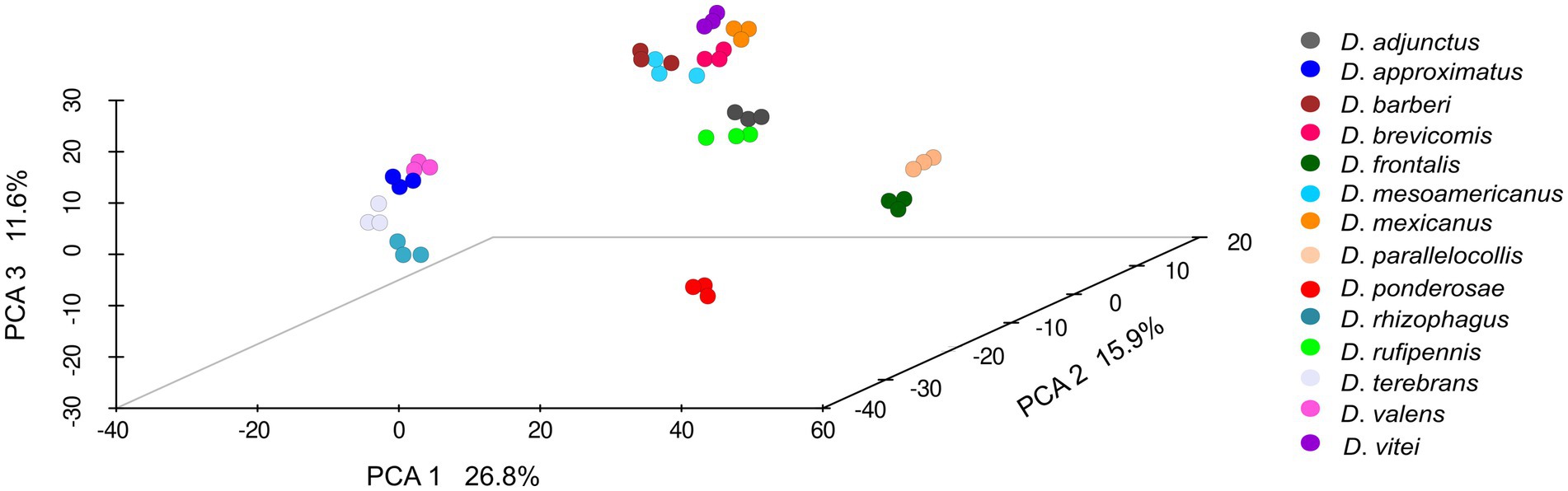
Figure 3. Principal component analysis (PCA, β-diversity) of the gut fungal assemblages from 14 species of Dendroctonus. (PERMANOVA, p < 0.05). Non-significant differences were found among within-species replicates (PERMDISP, p > 0.05). Dots of the same color represent biological replicates.
A relaxed CMC composed mainly of taxa in the genera Candida (present in all bark beetles, 100%), Nakazawaea (13 species; 92.86%), Cladosporium (10 species; 71.43%), Ogataea (10 species; 71.43%), and Yamadazyma (9 species; 64.28%) was recovered from the assemblages of Dendroctonus species (Figure 4). However, beetle species that shared the same fungal genera did not always share the same ASVs (Supplementary Table 1).
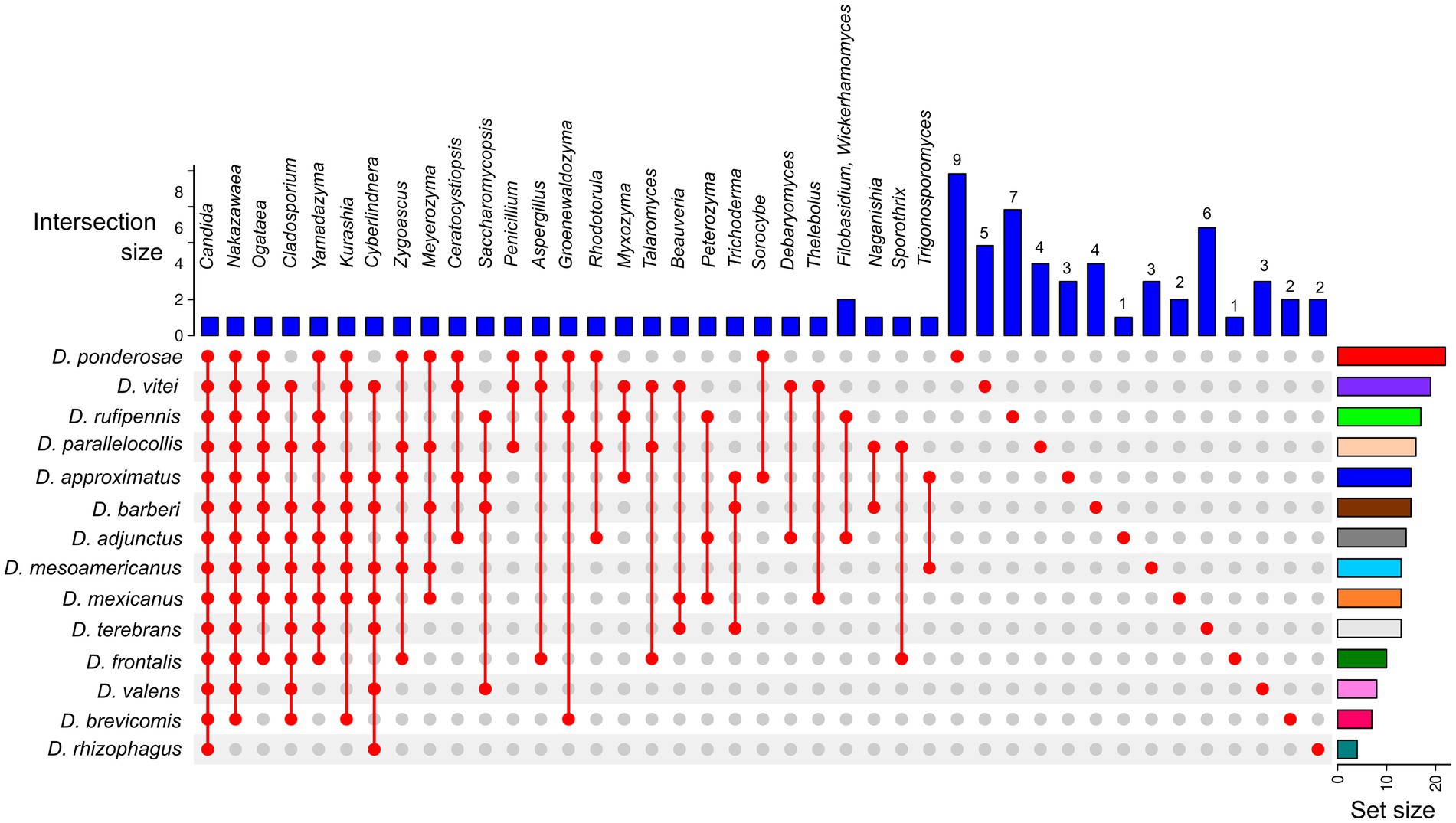
Figure 4. UpSetR plot of the core mycobiome from 14 Dendroctonus species. Colored bars (plot’s right side) highlight the number of total fungal genera present in each Dendroctonus species. Blue bars (plot’s top part) represent the number of fungal shared or single genera among beetle species. The red points (plot’s bottom) represent the presence or absence of fungal genera in the Dendroctonus species.
An analysis of the metabolic pathways of the CMC (Candida, Nakazawaea, Cladosporium, Ogataea, and Yamadazyma) shows that these fungi do not possess all enzymes involved in the degradation of plant cell walls. Cellulose is degraded exclusively by Cladosporium (EC 3.2.1.4 and EC 3.2.1.21), whereas glucan (EC 3.2.1.58) is degraded by all five members, and there is no evidence that these fungi degraded lignin or rhamnose (Supplementary Figure 3). On the other hand, core fungi can synthesize threonine (EC 2.7.1.39 and EC 4.2.3.1) but do not have genes encoding the biosynthesis of essential amino acids (Ile, Val, Leu, Trp, Phe, Lys, His, and Met). A total of six trophic modes including 14 different guilds were identified from 248 fungal ASVs. The frequency of these trophic modes varied among libraries and beetle species, with the most frequent being pathotroph-saprotroph-symbiotroph (47.41%), saprotroph-symbiotroph (35.62%), and saprotroph only (14.31%), whereas the less frequent was symbiotroph (1.48%), and other trophic modes had a frequency of less than 2%. ASV into these trophic modes were associated to animal pathogen, endophyte, endosymbiont, epiphyte, saprotroph, undefined saprotroph, soil and wood saprotroph, as well as animal or plant symbiotroph (Figure 5; Supplementary Table 2).
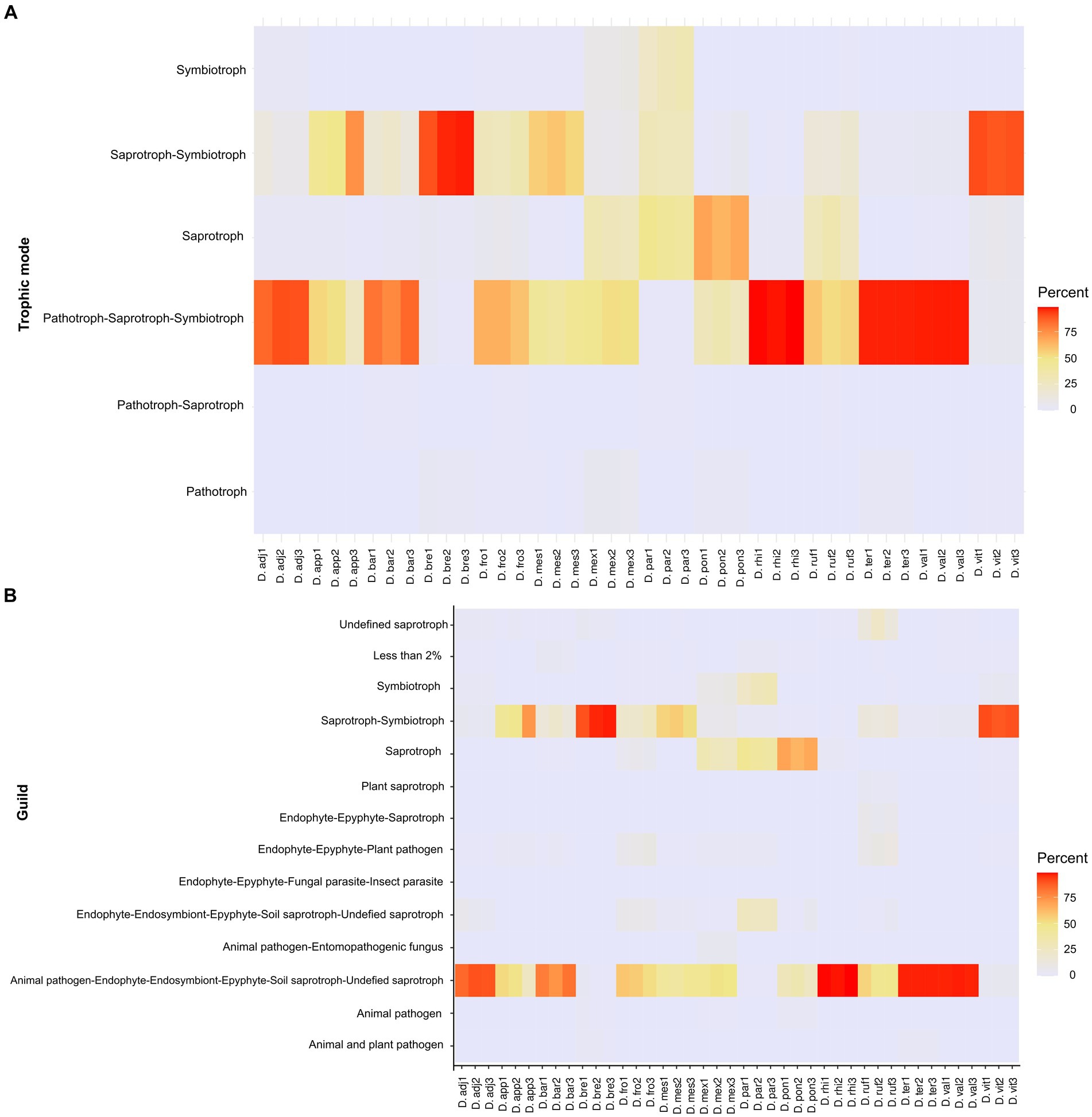
Figure 5. Heat maps showing relative frequencies of trophic modes (A) and guilds (B) predicted in each library of fungal assemblages, based on the FUNGuildR database.
3.3 Network analysis of fungal assemblagesFrom 248 fungal ASVs recovered, only 115 had >23 reads. The global network was integrated by four modules, which included 11 ASV nodes from seven filamentous fungi, and 43 ASV nodes from 10 yeasts (Figure 6). The 54 nodes were connected by 530 statistically significant edges, of which 291 were positive edges (i.e., mutualistic and communalistic interactions) and 239 were negative edges (i.e., antagonistic interactions) (Figures 6A,B). In the global network, there was no presence of hub nodes (nodes that have a low within-module connectivity, zi < 2.5, Pi > 0.3); however, nine of them were connectors (nodes with a high fraction of their links to other modules, zi < 2.5, Pi ≥ 0.62) and 45 peripherals (nodes with most links within their module and with other modules, zi < 2.5; Pi < 0.62). The ASV Candida Can_ore2 was the no-hub peripheral node with the highest level of edges, and the ASV Nakazawaea Nak_amb1 was the connector no-hub node with the highest connectivity and efficiency among all ASV interactions in the network. The global network was statistically significant to null random networks (z-score = 2.75, p < 0.003), and the connectivity distribution followed the power law model (R2 = 0.001). The values of modularity (M = 0.538), average connectivity (avgK = 19.63), average path distance (GD = 1.655), and average clustering coefficient (avgCC = 0.561) were significantly higher in the global network than the null random network values (M = 0.099 ± 0.010, GD = 1.644 ± 0.004, and avgCC = 0.484 ± 0.008).
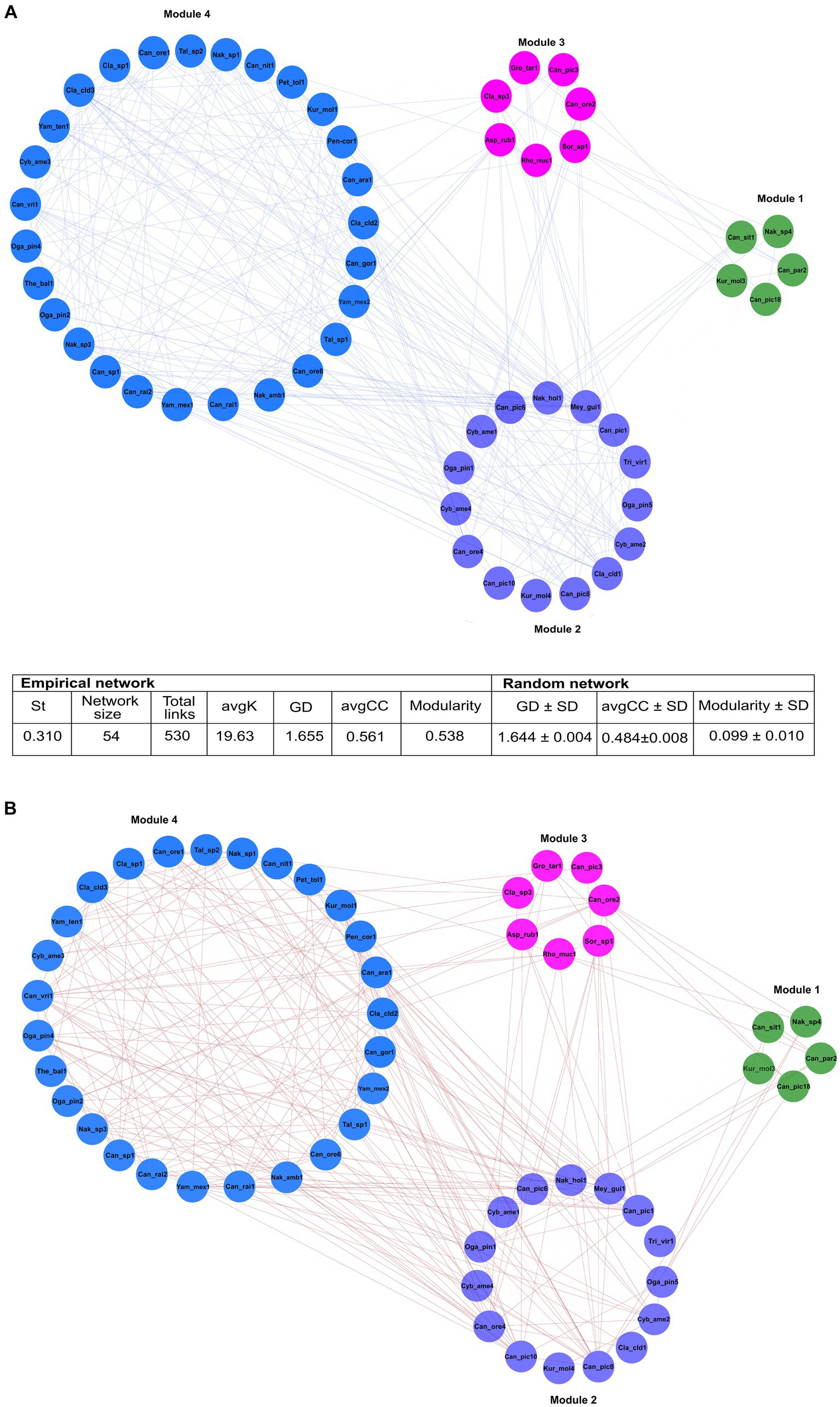
Figure 6. The general ecological networks of the fungal assemblages associated with 14 species of Dendroctonus genus. It was built with 115 ASVs whose reads were > 23. Four modules integrated in the general network, which included 11 ASV nodes from 7 genera of filamentous fungi, and 43 ASV nodes from 10 yeasts genera. The values of modularity, average connectivity, average path distance, and average clustering coefficient were significantly higher than the random network. Significant positive (A) or negative (B) correlations (p < 0.05) within the same and different modules were separated and represented by blue and red lines, respectively. Asp_rub (Aspergillus ruber); Can (Candida sp.); Can_ara (Candida arabinofermentans); Can_gor (Candida gorgasii); Can_nit (Candida nitratophila); Can_ore (C. oregonensis); Can_par (C. parapsilosis); Can_pic (C. piceae); Can_rai (C. railenensis); Can_sit (Candida sithepensis); Can_vri (C. vriesea); Cla (Cladosporium sp.); Cla_cld (C. cladosporioides); Cyb_ame (Cyberlindnera americana); Kur_mol (Kuraishia molischiana); Gro_tar (Groenewaldozyma tartarivorans); Mey_gui (Meyerozyma guilliermondii); Nak_sp. (Nakazawaea_sp.); Nak_amb (N. ambrosiae); Nak_hol (N. holstii); Oga_pin (Ogataea pini); Pen_cor (Penicillium corylophilum); Pet_tol (Peterozyma toletana); Rho_muc (Rhodotorula mucilaginosa); Sor_sp (Sorocybe sp.); Tal_sp (Talaromyces sp.); The_bal (Thelebolus balaustiformis); Tri_vir (Trichoderma viride); Yam_mex (Yamadazyma mexicana); and Yam_ten (Yamadazyma tenuis).
Module 1 comprised five ASVs in total: three of Candida (Can_par2, Can_sit1, Can_pic18), one of Nakazawaea (Nak_sp4), and one of Kuraishia (Kur_mol3) from four beetle species. These ASVs presented five positive and two negative interactions among them. Can_sit1 and Kur_mol3 ASVs connected module 1 with module 2, and Can_pic18, Can_par2, and Nak_sp4 with modules 2 and 3 (Supplementary Table 2).
Module 2 was formed by 15 ASVs in total: five of Candida (Can_ore4, Can_pic8, Can_pic10, Can_pic6, Can_pic1), one of Meyerozyma (Mey_gui1), two of Ogataea (Oga_pin1, Oga_pin5), three of Cyberlindnera (Cyb_ame1, Cyb_ame4, Cyb_ame2), one of Kuraishia (Kur_mol4), one of Nakazawaea (Nak_hol1), and two filamentous fungi of the genera Trigonosporomyces (Tri_vir1) and Cladosporium (Cla_cld1) from 13 beetle species. Within this module, ASVs had 43 positive and 14 negative interactions among them. Can_pic1, Can_pic10, Can_ore4, Cyb_ame2, and Cla_cld1 linked module 2 with modules 3 and 4; Can_pic1, Can_pic6, Can_pic8, Cyb_ame1, Oga_pin1, Nak_hol1, and Mey_gui1 linked it to modules 1, 3, and 4; Oga_pin5 connected it to modules 1 and 3; Kur_mol4 linked it to the modules 0 and 3; and Tri_vir1 associated it with the modules 2 and 3 (Supplementary Table 3).
ASVs of the yeasts Candida (Can_ore2 and Can_pic3), Groenewaldozyma (Gro_tar1), and Rhodotorula (Rho_muc1), and the filamentous fungi, Cladosporium (Cla_sp3), Aspergillus (Asp_rub1), and Sorocybe (Sor_sp1) from eight beetle species comprised module 3. These ASVs presented eight negative and 10 positive links among them within the module. Can_ore2, Can_pic3, Rho_muc1, Asp_rub1, and Cla_sp3 interconnected this module with the modules 1, 2, and 4, while Gro_tar1 and Sorocybe (Sor_sp1) linked it with the modules 1 and 2 (Supplementary Table 2).
Module 4 consisted of 23 ASVs of the yeasts Nakazawaea (Nak_sp1, Nak_amb1, and Nak_sp3), Candida (Can_ore1, Can_ore6, Can_rai1, Can_rai2, Can_sp1, Can_vri1, Can_gor1, Can_nit1, and Can_ara1), Peterozyma (Pet_tol1), Kuraishia (Kur_mol1), Yamadazyma (Yam_mex1, Yam_mex2, and Yam_ten1), Ogataea (Oga_pin2 and Oga_pin4), and Cyberlindnera (Cyb_ame3) and seven ASVs from the filamentous fungi Cladosporium (Cla_sp1, Cla_cld2, Cla_cld3), Penicillium (Pen_cor1), Talaromyces (Tal_sp1 and Tal_sp2), and Thelebolus (The_bal1) from 10 beetle species. These ASVs had 86 negative and 96 positive links between them within the module. Most of the ASVs were linked to modules 2 and 3, while Can_rai2, Can_gor1, Can_ore1, Can_ore1, Can_ore6, Can_nit1, Can_sp1, Yam_mex1, and Cla_sp1 linked it to module 2; Oga_pin2 connected it to module 2; Can_rai1 linked it to modules 1, 2, and 3; and Can_rai1 linked it to the modules 1, 2, and 3 (Supplementary Table 3).
The bipartite networks confirmed that Candida, Cyberlindnera, Yamadazyma, Cladosporium, Ogataea, and Nakazawaea were key genera that maintained the structure and stability of the community (modularity 0.38, nestedness 19.0, and connectedness 0. 65). However, a plethora of fungal genera of low frequency showed some degree of specialization in each beetle assemblages (modularity 0.27, nestedness 26.07, and connectedness 0.10) (Figure 7).
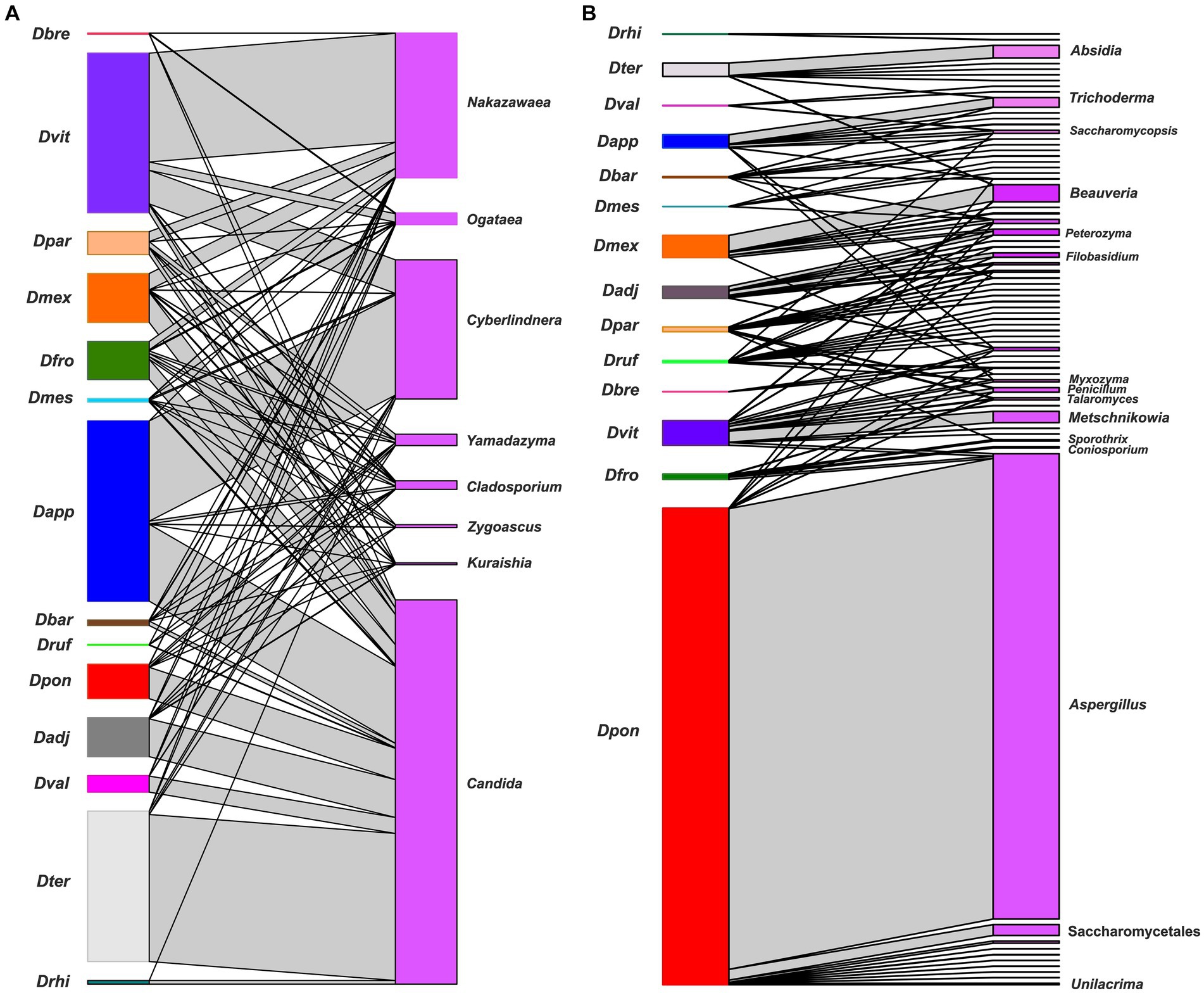
Figure 7. A bipartite network between Dendroctonus species and the gut fungal assemblages. Dendroctonus species are represented in the nodes on the left side and fungal ASVs in the nodes on the right side, the connections between insects and fungi are represented by gray connectors (or links). (A) The network of the most dominant fungal genera. (B) The network of non-dominant fungal genera. Dadj: D. adjunctus, Dapp: D. approximatus, Dbar: D. barberi, Dbre: D. brevicomis, Dfro: D. frontalis, Dmes: D. mesoamericanus, Dmex: D. mexicanus, Dpar: D. parallelocollis, Dpon: D. ponderosae, Drhi: D. rhizophagus, Druf: D. rufipennis, Dter: D. terebrans, Dval: D. valens, and Dvit: D. vitei.
4 DiscussionIn this study, we comprehensively characterized the composition and diversity of gut-associated fungi of Dendroctonus bark beetles using the ITS2 region and high-throughput sequencing technologies. Our results indicate that gut fungal assemblages of the Dendroctonus spp. are represented mainly by Ascomycetous fungi, which agrees with other reports from the bark and ambrosia beetles, as well as other Coleoptera groups (Harrington, 2005; Suh et al., 2005; Henriques et al., 2006; Rivera et al., 2009; Gao et al., 2018; Biedermann and Vega, 2020; Chakraborty et al., 2020, 2023; Ibarra-Juarez et al., 2020; Veselská et al., 2023).
At the genus level, α-diversity was integrated into 71 fungal genera including 32 yeast ASVs and 39 filamentous fungi ASVs. Differences in richness and diversity among Dendroctonus gut assemblages were driven mainly by filamentous ASVs from fungi and yeast with a relative abundance of <1%. However, our results confirm that yeasts are the most abundant group and persistent in the gut of these bark beetles, which matches earlier reports for Dendroctonus spp. using culture-dependent methods (Rivera et al., 2009) and with other studies carried out with bark beetles that find yeasts and yeast-like fungi are primary taxa in gut assemblages (e.g., Chakraborty et al., 2020).
Gut fungal assemblages of Dendroctonus beetles differed considerably in α-diversity, including taxonomic ASV richness and abundance. In general, Dendroctonus species that feed on roots or the lower bole or colonize trees non-lethally in their native ranges, such as D. rhizophagus, D. terebrans, and D. valens, exhibited the least diverse gut fungal assemblages (Figure 2). In contrast, highly “aggressive” or “primary” Dendroctonus spp. that are often a driving factor in tree death, including D. adjunctus, D. frontalis, D. mesoamericanus, and D. rufipennis, generally had more diverse gut fungal assemblages. However, metrics of α-diversity also varied strongly across biological replicates for some species (e.g., D. mexicanus, D. parallelocollis, and D. vitei), indicating a potential need for additional sampling within species or across regions to fully characterize community variation.
The dominant yeasts were Candida, Nakazawaea, Cyberlindnera, Yamadazyma, and Ogataea (Figure 1). The high prevalence of yeast in the gut indicates that they are potentially important for beetle development and nutrition. Two pathways exist by which yeasts may impact beetle nutrition. First, during development, beetles almost certainly directly consume yeast cells that proliferate in the subcortical gallery environment. To the best of our knowledge, no studies have yet tested variation in beetle development with and without access to gut yeasts, likely due to the difficulties in rearing Dendroctonus axenically. One recent study showed that crude P content, but not N content, of yeast cells was not strongly different from tree phloem (Davis et al., 2023), indicating that at least some yeast isolates are not substantially more protein-rich than tree tissues alone. However, yeasts also produce enzymes that may be involved in the degradation of structural and non-structural polysaccharide complexes including cellulose, lignin, or starch, which could alter the C:N ratios or reduce tissue toughness. For instance, Candida arabinofermentans converts L-arabinose, a common pentose in lignocellulose, to ethanol (Kurtzman and Dien, 1998); Candida nitratophila assimilate nitrates (Ali and Hipkin, 1986); and both Cyberlindnera americana and Candida piceae have lipolytic and amylolytic abilities (Briones-Roblero et al., 2017b).
Second, yeasts may alter nutritional opportunities indirectly via community-driven effects. For example, some yeasts (e.g., Ogataea) can alter the growth of filamentous fungal symbionts (Paine and Hanlon, 1994; Davis et al., 2011), while others (e.g., Yamadazyma) in some cases inhibit it (Wang et al., 2013; Davis et al., 2023; González-Gutiérrez et al., 2023). In addition, some yeast taxa (e.g., Kuraishia, Ogataea, and Nakazawaea) degrade terpenes and/or affect pheromone production (Hunt and Borden, 1990), and several in silico studies in yeasts (e.g., Candida and Cyberlindnera) demonstrate the presence of genes and transcripts related to terpene detoxification, such as cytochromes P450, and Transporters ABC, MATE, and MFS, and others (Hernández-Martínez et al., 2016; Soto-Robles et al., 2019).
The low abundance recorded in the gut of the fungal symbionts, including Ceratocystiopsis, Entomocorticium, Ophiostoma/Sporothrix, and Grosmannia/Leptographium, is surprising and consistent with other culture-based and culture-independent studies (Rivera et al., 2009; Bozorov et al., 2019), confirming that these symbionts are primarily associated with mycetangia (Vazquez-Ortiz et al., 2022) or body surfaces (Gao et al., 2018; Chakraborty et al., 2020) from Dendroctonus bark beetles. It is generally regarded as canon that Dendroctonus beetles feed on their respective associated mycetangial fungal taxa during development (Six and Klepzig, 2004; Six and Wingfield, 2011); however, it is puzzling why these taxa (Entomocorticium, Ophiostoma, Grosmannia) were not abundant in the gut if this is the primary nature of the association.
There are several possible explanations for this pattern. First, insect digestive tracts contain microenvironmental variation in oxic and anoxic gradients (Ceja-Navarro et al., 2014), which may limit the proliferation of mycetangial fungi in the gut. Second, mycetangial fungi may be rapidly digested by beetles or degraded by joint action of chitinases present in both bacteria and yeasts (Fabryová et al., 2018; Bozorov et al., 2019; Ibarra-Juarez et al., 2020; Peral-Aranega et al., 2023). Third, presumably mycetangial fungi are fed upon primarily during larval development, which would lead us to assume that they should be present at this stage of development. However, these filamentous fungi are not isolated from the gut of Dendroctonus larvae using culture-dependent methods (Rivera et al., 2009). Even so, more detailed studies are required to confirm that larvae may have abundances of very different gut fungal assemblages than adult Dendroctonus, because individuals sampled in the present study were adults.
Instead, the dominant filamentous fungus in the alimentary canal was Cladosporium (present in 71% of beetle species), with approximately equivalent representation across species of Aspergillus, Beauveria, Ceratocystiopsis, and Trichoderma (present in ~21–28% of beetle species). It is unclear which of these associations are truly “symbiotic” relationships and which are incidental or even exploitative. Beauveria is an important entomopathogen for bark beetles and insects in general (Mann and Davis, 2021); similarly, some species of Aspergillus are pathogenic to bark beetles (Zeiri et al., 2017), though both fungi are relatively ubiquitous in natural environments (Ramirez-Camejo et al., 2012; Maistrou et al., 2020). Their presence in the gut of several species may indicate potential fungal parasitism in sampled populations. The ecological relationships between Cladosporium spp. and bark beetles are less clear; though some Cladosporium isolates may elevate the N content of plant tissues (Solomon and Oliver, 2001) or participate in the lignin degradation by the presence of laccases (Aslam et al., 2012). Cladosporium is isolated from various bark beetle species in many different geographic regions (Bateman et al., 2016; Davydenko et al., 2017). Cladosporium spp. are also environmentally ubiquitous on plant surfaces (Katsoula et al., 2021) but may also be endophytes (Ganley and Newcombe, 2006), plant pathogens (De Wit et al., 1997), entomopathogens (Samways and Grech, 1986), and even parasites of other fungi (Torres et al., 2017). Since Cladosporium spp. are represented in the gut of >70% of Dendroctonus species, further exploration of their role in nutrition and interactions with beetles and host trees is warranted.
On the other hand, results show a different β-diversity when guts among beetle species were compared (Figure 3; Supplementary Figures 2A,B), without the influence of the intraspecific variation of assemblages of each insect species. Different metrics indicate that each beetle species was generally associated with a relatively distinct gut fungal assemblage. The spatial disjunction of fungal assemblages (PCA) indicates a strong replacement of fungal ASVs, though there was also overlap in assemblages across species (Figure 4; Supplementary Figures 2A,B). Differences are given both by fungal ASVs with high and low abundance of shared genera between assemblages, as well as by unique or rare ASVs commonly of low abundance.
Although Dendroctonus spp. surveyed here were associated fully with the genus Candida and most with Nakazawaea, Ogataea, and Yamadazyma, our results show a core mycobiome integrated by different Candida ASVs (Figures 4, 5), which significantly contribute to the structure and stability of the community. This also suggests that beetles are often associated with cryptically diverse and functionally redundant assemblages of gut symbionts with potentially complementary enzymatic capabilities, to the benefit of all, including the insect host. Furthermore, this also suggests that the maintenance of fundamental functions across the incorporation or retention of symbionts with complementary capabilities or redundant is a viable strategy, which favors all interaction types among fungal symbionts as suggested by our ecological and bipartite networks, rather than the retention of strict specific symbionts.
The observed differences in both diversities may be due to different factors, such as biological (e.g., the type of life cycle and its duration and generation number), ecological (e.g., tree host species, distribution range both of insects and host, colonization strategy, endemic or epidemic condition of populations, and interaction with phoretic ectosymbionts or with other arthropods) and morpho-functional (e.g., gut compartmentalization and physicochemical microenvironment), which are dynamic in space and time and non-mutually exclusive. Furthermore, some of these factors can be influenced by the diet and the subcortical environment (Romeralo et al., 2022; Chakraborty et al., 2023; Ge et al., 2023), despite these insects exploiting a similar resource in terms of nutrition and habitat. Given that we lack a general understanding of how these factors affect mycobiome diversity (Rivera et al., 2009; Davis, 2015; Gao et al., 2018), in part because it is regulated by overlapping processes on spatial and temporal scales ranging from the global to the local, future studies with specific experimental designs are required to test the individual influence of different drivers.
The multiple positive or negative associations observed in the overall network suggest that fungal ASVs establish intimate interactions among them, which may be cooperative or mutualistic, as they contribute to the processes of terpene detoxification,
留言 (0)