
記住我





Adult male Sprague–Dawley rats weighing 250–300 g were purchased from SPF Biotechnology Co., Ltd. (Beijing, China). The rats were housed under a 12/12 h light/dark cycle at 23 ± 1 ℃ with free access to food and water. The Animal Care and Use Committee of China Medical University approved all procedures used in the study (IACUC no. CMU2022004).
NP model inductionThe CCI rat model of NP was established according to procedures described by Bennett and Xie [33]. Firstly, rats were anesthetized with 1% isoflurane, and a 1.5 cm lateral incision was made in the right hindlimb. The muscle was bluntly separated to expose the sciatic nerve trunk. Secondly, the sciatic nerve trunk was loosely ligated by four silks (4–0#) at an interval distance of 1 mm. The sutures were gently tightened until a brisk twitch in the right hindlimb was observed. Finally, layered skin suturing was performed.
Mechanical allodynia (von Frey test)Mechanical allodynia was assessed by measuring paw withdrawal threshold (PWT) in response to von Frey filament stimulation. The rats were placed in a transparent plastic cage with a mesh bottom for 20 min before testing. PWT was assessed using a dynamic plantar esthesiometer (Ugo Basile, 37,450, Italy). A probe was applied to the mid-plantar surface of the hind paw under increasing pressure. The cut-off pressure was set to 50 g, and the esthesiometer automatically recorded the force causing the withdrawal response. For each animal, ≥ 3 measurements were performed every 10-min to stimulate each hind paw.
Thermal hyperalgesia (hot plate test)Thermal hyperalgesia was assessed by measuring paw withdrawal latency (PWL) in response to hot plate test. A hot plate analgesia meter (Ugo Basile, 35,300, Italy) was prepared with a pre-set plate temperature of 53 ± 1 °C. Once the rat was placed on the hot plate, the time between placement and licking, shaking, or stepping of the hindpaws was recorded. The cut-off time was set to 30 s to avoid tissue damage. For each animal, ≥ 3 measurements were performed every 10 min.
Drugs and administrationThe effective and selective MMP-2 inhibitor, ARP 100 (MMP-2 inhibitor III, compound 10a), was purchased from Selleck Chemicals (Shanghai, China). The MMP-2 inhibitor was dissolved in dimethyl sulfoxide (DMSO) to a concentration of 1 nmol/µL. The doses of MMP-2 inhibitor were chosen as previously described [30]. The in vivo SPOCK2-siRNA (2 Ome + 5 Chol) was commercially synthesized by RiboBio Co. Ltd. (Guangzhou, China). The specific target sequence of SPOCK2-siRNA was 5′-GTGAGAACTCGAAGCAGAA-3′. Negative control siRNA (NC-siRNA) was synthesized using a scrambled sequence of nucleotides. After methylation and cholesterol modification, in vivo siRNA can be stably transfected in vivo and have high transfection efficiency without transfection reagents [34].The freeze-dried siRNA powder was dissolved in 0.9% normal saline (NS). The method of administration was chosen as previously described [35]. The recombinant lentiviral vectors carrying SPOCK-shRNA (pGLV-3-GFP-SPOCK2-shRNA) was purchased from GenePharma Co. Ltd. (Shanghai, China). The target sequence of SPOCK2-shRNA was 5′-GUGAGAACUCGAAGCAGAA-3′, and the negative control shRNA vector with a scrambled shRNA (shScr) insert was used to control any effects caused by non-RNAi mechanisms. The recombinant SPOCK2 protein (rSPOCK2) was purchased from R&D Systems (Minneapolis, MN). The rSPOCK2 was dissolved in 0.9% NS to a concentration of 0.1 ng/µL. The doses and administration of rSPOCK2 were based on previous literature [8]. Drugs or vehicles (DMSO, NS) were delivered into the cerebrospinal fluid space around the lumbosacral spinal cord through intrathecal (i.t.) administration.
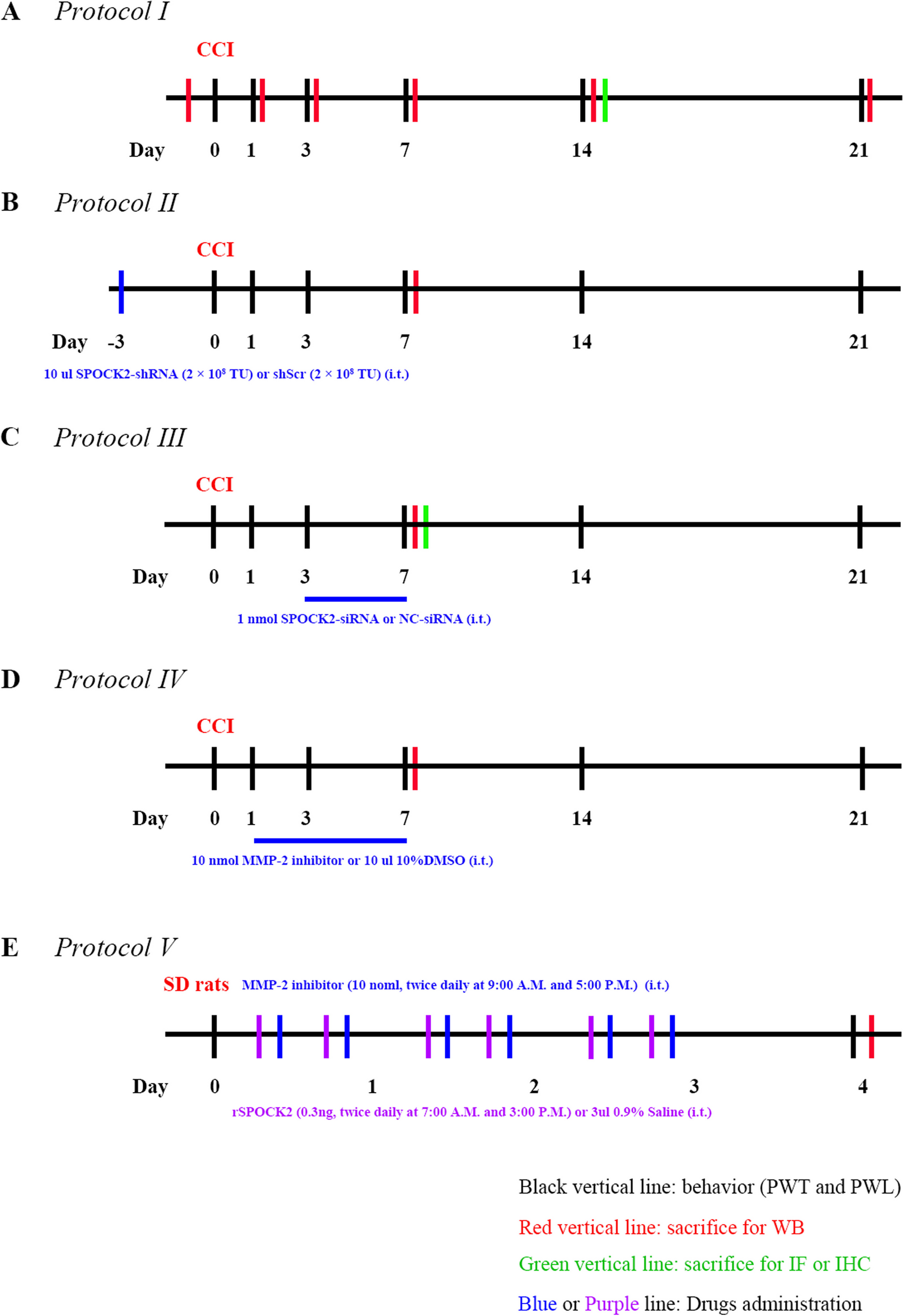
Experimental protocolsProtocol ITo test the time course of changes in pain behaviors and expression of SPOCK2 in the spinal cord, rats were assigned to sham (n = 5) and CCI (n = 30) groups. The CCI group underwent CCI surgery, while the same procedure was performed in the sham group, except for the ligation of the sciatic nerve. PWT and PWL were assessed at the ipsilateral hind paws 24 h prior to CCI and day 1, 3, 7, 14 and 21 after CCI. Sham rats were sacrificed at final time point and five CCI rats were sacrificed after PWT and PWL measurement at each time point (Fig. 1A). The protein was extracted from L4-6 rat spinal cord of ipsilateral side for western blot analysis. Additional CCI rats used for immunofluorescence and immunohistochemistry were sacrificed at 14 days after CCI.
Fig. 1
Schematic illustration of the experimental protocols
Protocol IITo explore the role of SPOCK2 in the spinal cord in the process of pain occurrence, rats were divided into 4 groups (n = 5 for each group): sham; CCI; CCI treated with shScr (CCI + shScr); CCI treated with SPOCK2-shRNA (CCI + SPOCK2-shRNA). 10ul SPOCK2-shRNA (2 × 108 TU) or 10ul scrambled shRNA (2 × 108 TU) was i.t. injected 3 days prior to the beginning of CCI. PWT and PWL were assessed at the ipsilateral hind paws 24 h prior to CCI and day 1, 3, 7, 14 and 21 after CCI, and rats were sacrificed at the end of the protocol. Another four groups (n = 5 for each group) rats used for western blot analysis were sacrificed at 7 days after CCI (Fig. 1B).
Protocol IIITo further examine the role and mechanism of SPOCK2 in the development of NP, rats were divided into 4 groups (n = 5 for each group): sham; CCI; CCI treated with NC-siRNA (CCI + NC-siRNA); CCI treated with SPOCK2-siRNA (CCI + SPOCK2-siRNA). 1 nmol SPOCK2-siRNA or NC-siRNA was i.t. injected 5 continuous days (after CCI, 3–7 days). PWT and PWL were assessed at the ipsilateral hind paws 24 h prior to CCI and day 1, 3, 7, 14 and 21 after CCI, and rats were sacrificed at the end of the protocol. Another four groups (n = 5 for each group) rats used for western blot analysis were sacrificed at 7 days after CCI. Additional CCI rats treated with NC-siRNA and SPOCK2-siRNA used for immunofluorescence were sacrificed at 7 days after CCI (Fig. 1C).
Protocol IVTo further study the relationship between SPOCK2 and MMP-2 in the development of NP, rats were divided into 4 groups (n = 5 for each group): sham; CCI; CCI treated with 10%DMSO (vehicle) (CCI + 10%DMSO); CCI treated with MMP-2 inhibitor (CCI + MMP-2 inhibitor). 10ul 10%DMSO or 10 nmol MMP-2 inhibitor was i.t. injected 7 continuous days (after CCI, 1–7 days). PWT and PWL were assessed at the ipsilateral hind paws 24 h prior to CCI and day 1, 3, 7, 14 and 21 after CCI, and rats were sacrificed at the end of the protocol. Another four groups (n = 5 for each group) rats used for western blot analysis were sacrificed at 7 days after CCI (Fig. 1D).
Protocol VTo further study the role of SPOCK2 involved in NP and the relationship between SPOCK2 and MMP-2 in the development of NP, rats were divided into 3 groups (n = 5 for each group): SD rats treated with 0.9% NS (Vehicle); SD rats treated with rSPOCK2 (rSPOCK2); SD rats treated with rSPOCK2 and MMP-2 inhibitors (rSPOCK2 + MMP-2 inhibitor). We i.t. injected rSPOCK2 (0.3 ng, twice daily at 7:00 A.M. and 3:00 P.M.) or 3ul 0.9%NS (Vehicle) for 3 days. In the rSPOCK2 + MMP-2 inhibitor group, MMP-2 inhibitor was i.t. injected (10 nmol, twice daily at 9:00 A.M. and 5:00 P.M.) for 3 days. 16 h (day 4) after the end of drug administration, PWT and PWL were evaluated. Rats used for western blot analysis were sacrificed at the end of the protocol (Fig. 1E).
Western blotIsolated rat lumbar spinal cord (L4–6) was homogenized in an ice-cold lysis buffer containing a protease and phosphatase inhibitor cocktail. Supernatants were collected by centrifugation at 12,000 ×g for 15 min at 4 °C. Total protein was obtained from the spinal cord using protein extraction kits, separated by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and transferred onto polyvinylidene difluoride (PVDF) membranes. The membranes were next placed in blocking buffer (5% milk in Tris-buffered saline with Tween-20) for 1 h and incubated overnight (16–18 h) at 4 °C with primary antibodies against SPOCK2 (1:1,000, Cat# 217,044, RRID: AB_3076329, Abcam, Cambridge, UK), MT1-MMP (1:1,000, Cat# 14,552–1-AP, RRID: AB_2250751, Proteintech, Wuhan, China), MMP-2 (1:1,000, Cat# 66,366–1-Ig, RRID: AB_2881746, Proteintech), ERK1/2 (1:2,000, Cat# 9102, RRID: AB_330744, Cell Signaling Technology, Danvers, MA, USA), p-ERK1/2 (1:1,000, Cat# 4370, RRID: AB_2315112, Cell Signaling Technology), interleukin-1β (IL-1β) (1:1,000, Cat# sc-7884, RRID: AB_2124476, Santa Cruz, TX, USA), CC-chemokine ligand 2 (CCL2) (1:2000, Cat# ab7202, RRID: AB_305755, Abcam), JNK (1:3,000, Cat# 17,572–1-AP, RRID: AB_2266214, Proteintech), p-JNK (1:2,000, Cat# 80,024–1-RR, RRID: AB_2882943, Proteintech), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (1:10,000, Cat# 60,004–1-Ig, RRID: AB_2107436, Proteintech). After washing three times with TBST for 5 min, the membranes were incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-rabbit, 1:10,000, Cat# SA00001-2, RRID: AB_2722564, Proteintech) (anti-mouse, 1:10,000, Cat# SA00001-1, RRID: AB_2722565, Proteintech) for 1 h at room temperature. The labeled proteins were visualized and quantified using an enhanced chemiluminescence detection system.
ImmunofluorescenceFrozen immunofluorescence sections were used for these experiments. Briefly, rats were transcardially perfused with 0.9% normal saline (NS), followed by 4% paraformaldehyde. Spinal cords were dissected, fixed overnight in 4% paraformaldehyde at 4 °C, and then continuously dehydrated in 20% and 30% sucrose for 24 h each. Spinal cord sections were sliced 10-µm thick with a cryostat. Following non-specific binding with 5% goat serum in 0.3% Triton for 1 h at room temperature, the sections were incubated overnight (16–18 h) at 4 °C with primary antibodies against SPOCK2 (1:200, Cat# 217,044, RRID: AB_3076329, Abcam), MT1-MMP (1:200, Cat# 14,552–1-AP, RRID: AB_2250751, Proteintech), MMP-2 (1:200, Cat# 66,366–1-Ig, RRID: AB_2881746, Proteintech), glial fibrillary acidic protein (GFAP) (1:400, Cat# 3670, RRID: AB_561049, Cell Signaling Technology), and monoclonal mouse anti-RAT CD11b (1:400, Cat# MCA275R, RRID: AB_321302, BIO-RAD, USA). On the second day, the sections were incubated for 1 h at room temperature with the following secondary antibodies: Alexa Fluor 488 goat anti-rabbit (1:400, Cat# A-11008, RRID: AB_143165, Thermo Fisher Scientific, USA) and Alexa Fluor 568 goat anti-mouse (1:400, Cat# A-11031, RRID: AB_144696, Thermo Fisher Scientific). A confocal microscope (FV3000, Olympus, Japan) was used to capture all images. Quantitative immunofluorescence analysis was calculated by Image J Software (National Institutes of Health, Bethesda, USA). The positive cells were counted in a 500 um × 500 um measuring frame. Cell counts were then used to determine the total number of positive cells per square millimeter.
ImmunohistochemistryImmunohistochemical paraffin sections were used. Briefly, rat spinal cords were isolated following transcardial perfusion in 0.9% NS and 4% paraformaldehyde, post-fixed, and embedded in paraffin. Spinal cord 4 µm-sections were obtained and next were deparaffinized in xylene and rehydrated in graded ethanol. After the non-specific binding was blocked with 5% goat serum in 0.3% Triton for 1 h at room temperature, the sections were incubated overnight (16–18 h) at 4 °C with primary antibodies against SPOCK2 (1:200, Cat# 217,044, RRID: AB_3076329, Abcam). The signal was visualized using Elivision Super HRP IHC Kits (Maixin-Bio, China) and 3,3-diaminobenzidine (Maixin-Bio, China), and the nuclei were counterstained with hematoxylin.
Cell culture and plasmids transfectionHEK-293 T cells were purchased from the Shanghai Cell Bank (GNHu17, Shanghai, China) and cultured in high-glucose Dulbecco’s modified Eagle’s medium (Invitrogen, 11,960,044) with 10% fetal bovine serum (FBS; FB15015; Clark Biosciences, USA), 1% Glutamax (Invitrogen, 35,050,061), and 1% Sodium Pyruvate 100 mM Solution (Invitrogen, 11,360,070) at 37℃ and 5% CO2.
The wild-type (WT) plasmid (pCMV6-SPOCK2-Myc-DDK) and control (empty) plasmid (pCMV6-Myc-DDK) were purchased from OriGene (Rockville, MD, USA). ΔKAZAL (mut1) plasmid (pCMV6-ΔKAZAL-myc-DDK), ΔSPARC_EC (mut2) plasmid (pCMV6-ΔSPARC_EC-myc-DDK), and ΔΔ (mut3) plasmid (pCMV6-ΔΔ-myc-DDK), containing both knockout parts of the ΔKAZAL and ΔSPARC_EC plasmids, were obtained from TSINGKE Biological Technology (Beijing, China). Lipofectamine 3000 (Invitrogen, Waltham, MA, USA) was used for plasmid transfection according to the manufacturer’s instructions.
Coimmunoprecipitation (Co-IP)HEK-293 T cells were plated in 10 cm dishes. When 90% confluency was reached, the cells were lysed for the assays. Co-IP assays were performed with an Immunoprecipitation Kit consisting of Protein A + G Magnetic Beads (P2179S, Beyotime Biosciences) according to manufacturer’s instructions.
Data analysisData analyses were performed using the GraphPrism 9.0 software (Graph Pad Software, San Diego, CA, USA). All data are presented as mean ± SEM. Image J Software (National Institutes of Health, Bethesda, USA) was used to process the density of the western blot bands and quantitative immunofluorescence analysis. The normal distribution of data was analyzed using the D’Agostino and Pearson test (p > 0.05). Correlation was analyzed using the Pearson correlation test. Differences between groups were compared by a one-way or two-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test. Statistical significance was set at *p < 0.05 or **p < 0.01.
留言 (0)