Fibroblast activation protein-α (FAP) is a transmembrane glycoprotein that is restrictively expressed on normal fibroblasts possesing a resting phenotype, but is overexpressed on the cell surface of activated stromal fibroblasts of most (>90%) human primary and metastatic carcinomas (breast, lung, colorectal etc.) [[1], [2], [3], [4]]. FAP's proteolytic activity is considered to play a crucial role in tumor growth, and is mediated through invasion and metastasis [5]. Its constrained tissue expression combined with its involvement in tumorigenesis makes it a highly encouraging pan-tumor marker for the development of targeted radiopharmaceuticals for imaging and therapy of carcinomas.
In this regard various antibodies [6,7], CAR T-cells [8,9], FAP vaccines [10], and most impressively cyclic peptides [11], peptidomimetics or small-molecule based inhibitors [5,[12], [13], [14], [15]], have been explored as strategies to target FAP. Most of the small-molecule based FAP inhibitors are mainly 2-cyanopyrrolidine (cyanoPro) derivatives. The nitrile group present in 2-cyanopyrrolidine-based inhibitors is the most widely utilized warhead due to its electrophilic nature and inherent stability. Jansen et al. investigated the effect of replacing nitrile moeity in N-(4-quinolinoyl)-Gly-(2-cyanopyrrolidine) scaffold either with a stronger electrophilic group such as boronic acid or an irreversibly binding chloromethyl ketone moiety. While the latter inhibited FAP with a low binding affinity (177 ± 5 nM), the non-difluorinated 2-pyrrolidinylboronic acid (boroPro) derivative showed FAP inhibitory potency almost comparable to its difluorinated nitrile analogue (3.7 ± 0.2 nM vs 3.2 ± 0.4 nM). This is noteworthy because in the same work, they highlighted the importance of having a 4,4-difluoro substituent on the (2S)-2-cyanopyrrolidine ring to allow enhanced FAP inhibition [[12], [13], [14]].
Contrary to the earlier notion that FAP exhibits an absolute requirement for glycine at P2 position (Berger and Schechter's nomeclature [16]), a few groups including ours, have shown that FAP can also tolerate a P2-d-alanine in addition to a P2-glycine [[17], [18], [19]]. [99mTc]Tc-iFAP ([99mTc]Tc-HYNIC-D-Ala-boroPro) [20] and [68Ga]Ga-, [177Lu]Lu- and/or [225Ac]Ac-PNT6555 [21] were recently reported as P2-d-alanine bearing radiotracers for SPECT imaging and as a PET theranostic pair, respectively (Fig. 1). Moreover, our previous findings suggest that a combination of P2-glycine and 2-pyrrolidinylboronic acid at P1 position may not be ideal for FAP-targeting either due to lack of stability or for reasons not yet fully understood. Thus, the presence of a d-alanine residue is more suitable in the context of boroPro-based FAP inhibitors.
The well known FAP inhibitor (FAPI)-based radiotracers [[22], [23], [24]] were obtained by expanding on Jansen et al.'s N-(4-quinolinoyl)-Gly-(2-cyanopyrrolidine) scaffold [14,25], with gallium-68 (68Ga)–labeled FAPI-04 (Fig. 1) being the most widely exploited FAP tracer in clinical trials to date [26]. The high tumor-to-background uptake ratio (TBR) and rapid renal clearance makes radiolabeled FAPIs (particularly, FAPI-02 and -04) ideal for cancer diagnosis. However, therapeutic value of most radiolabeled FAPIs is compromised due to their rapid tumor washout resulting in ineffective tumor radiation absorbed dose. Several strategies are being explored to enhance their tumor residence time. This includes, but is not limited to one of the following: (1) Incorporation of an albumin binder moiety into the radioligand thereby enhancing its blood circulation time [[27], [28], [29], [30], [31]]; (2) Multimerization or bifunctionalization of a high-affinity ligand to increase its FAP avidity such as BiOncoFAP [32], DOTA-2P(FAPi)2 [33], DOTA-4P(FAPi)4 [34], DOTA/DOTAGA.(SA.FAPi)2 [35,36], ND-bisFAPI [37], FT-FAPI [38] etc; (3) Use of cyclic peptides such as FAP-2286 (Fig. 1) [11,39]; and (4) Chemical modification of FAPI-04 framework to generate new inhibitors, of which FAPI-46 (Fig. 1) has particularly emereged as a top performing candidate [23].
Amongst these, FAPI-46 (leading candidate from the FAPI series) and FAP-2286 (cyclic peptide) are currently two of the leading FAP-targeted therapeutic candidates which have been shown to display durable tumor accumulation. In the preclinical study published by Osterkamp et al. a significantly higher retention in HEK-FAP tumor xenografts was reported for 177Lu-labeled FAP-2286 (21.1 %I.A./g at 3 h and 16.4 %I.A./g at 72 h post-injection (pi)), when compared to [177Lu]Lu-FAPI-46 (15.3 %I.A./g at 3 h and 1.6 %I.A./g at 72 h) head-on over time [11]. Noteworthy enough, Millul et al. carried out a comprehensive head-to-head comparative analysis of several different classes of FAP radioligands including the ones containing albumin binding motifs, to understand their durability and potential for therapeutic applications with long-lived isotopes like 177Lu [27]. While the trend that they observed for [177Lu]Lu-FAPI-46 derivatives in both low (HT1080.hFAP) and high (HEK293.hFAP) FAP-expressing tumor models was identical to those of previous reports, the tumor uptake for [177Lu]Lu-FAP-2286 in the high FAP-expressing HEK293.hFAP tumor model was found to plunge from 22.99 %I.A./g at 4 h to 4.05 %I.A./g at 72 h pi. Although [177Lu]Lu-FAP-2286 exhibited highest tumor uptake and retention resulting in superior tumor radiation absorbed dose with high tumor-to-background uptake ratios for most critical organs except kidneys, the tumor uptake was notably found to be FAP-expression dependent. This was exemplified by very low tumor uptake (3.42 %I.A./g at 4 h pi) of [177Lu]Lu-FAP-2286 in the low FAP-expressing HT1080.hFAP tumor model and the tumor uptake dropped rapidly over time (0.64 %I.A./g at 72 h pi). These discrepancies make it difficult to conclusively state the superiority of this currently leading cyclic peptide, re-opening gates for further modifications and/or exploitation of newer FAP-targeting constructs like the ones described in the current work.
Deducing from a fairly exhaustive series of aromatic and heteroaromatic compounds investigated for their FAP-targeting potential by various groups to date, it is obvious that the most potent ones possess either a monocyclic (eg. benzene or pyridine-based) or a bicyclic heteroaromatic (quinoline-based) P3 position residue [13,14,18,25,40]. Furthemore, the monocyclic ones have been shown to be less potent than bicyclic quinoline-based molecules [13,14,18,25,40]. In our understanding, no FAP-targeted ligand possessing a tricylic heteroaromatic P3 moiety has ever been described to date. This prompted us to investigate if incorporating a tricyclic heteroaromatic benzo[h]quinoline residue at the P3 position of the N-(4-quinolinoyl)-Gly-(2-cyanopyrrolidine) scaffold (quinoline → benzo[h]quinoline) could be a potential FAP-targeted pharmacophore.
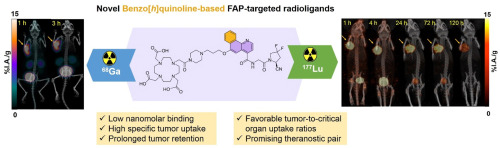
In this paper, we report the design, synthesis, and preclinical evaluation of two benzo[h]quinoline-based FAP-targeted constructs either containing N-(4-benzo[h]quinolinoyl)-Gly-difluoro-cyanoPro (SB03178) or N-(4-benzo[h]quinolinoyl)-D-Ala-boroPro (SB04033) scaffold (Fig. 2). Both the constructs were conjugated with 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) chelator, and labeled with natGa and 68Ga for in vitro FAP-inhibition assays, and PET imaging and ex vivo biodistribution analysis in an FAP-overexpressing HEK293T:hFAP xenograft model, respectively. 177Lu-labeled analogue of the outperforming candidate was subjected to multi-time point SPECT imaging, ex vivo biodistribution analysis, preclinical determination of tracer kinetics for dosimetry, with estimation of whole-body radiation absorbed dose to humans. The findings from this work enabled us to ascertain the potential of [68Ga]Ga/[177Lu]Lu-SB03178 as a theranostic pair due to its ability to share the same biological target along with different emission properties (positrons for PET imaging by 68Ga-labeled analogue, and cytotoxic β− particles along with γ-photons for radioligand therapy and SPECT imaging by the 177Lu-labeled analogue).

留言 (0)