Many biological surfaces are densely covered with a layer of carbohydrates, the so-called glycocalyx [1]. Recognition of these cell surface carbohydrate structures is accomplished by specialized lectins, constituting an important factor in cell–cell adhesion, pathogen recognition, and immune signaling processes. As a consequence, the pharmacological modulation of disease-relevant carbohydrate recognition events by means of glycomimetic ligands represents a promising drug design approach [2,3].
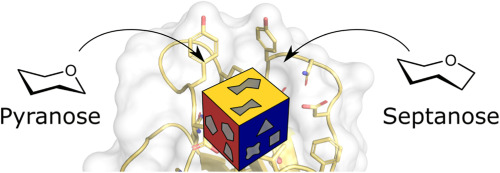
In terms of physicochemical and pharmacokinetic properties, the development of glycomimetic drugs is associated with a number of particular challenges. Specifically, low bioavailability and short plasma half-lives as well as rapid metabolic turnover of glycosidic bonds by glycosidases hamper glycomimetic drug design [2,3]. The design of drug molecules mimicking other classes of biomolecules has similarly been challenged. Thus, degradation of peptidic drugs or nucleotide-based therapeutics by proteases or nucleases, respectively, has historically been a major concern [[4], [5], [6], [7]]. One strategy to prevent metabolic lability focused on the incorporation of chemically disparate peptide or nucleotide analogs imitating the features of the natural building blocks but do not show their metabolic instability. Thus, modern strategies towards peptidic compounds include the use of peptide bond mimics such as β-amino acids or d-amino acid residues to prevent or delay proteolytic digestion [4,5,8,9]. Similarly, modification of oligonucleotides with phosphothioate backbones or incorporation of non-natural nucleobases such as N-methyl pseudouridine can efficiently prolong the half-life of RNA therapeutics [6,7,10]. For carbohydrates, similar efforts have mainly focused on modifying the glycosidic bond. Thus, researchers have attempted to replace O-glycosidic linkages with S-, N-, or C-glycosides in order to prevent glycosidase cleavage [3,[11], [12], [13], [14]]. The synthesis and application of such glycomimetics have been reviewed recently [15,16]. Inspired by the use of β-amino acids in peptide analogs, we sought to explore the compatibility of septanoses, ring-expanded homologs of pyranoses that are typically not recognized by glycosidases [17] with common design principles in glycomimetic drug discovery. For this, the bacterial adhesin FimH was selected as a well-characterized lectin target with abundant structure–activity relationship data [[18], [19], [20], [21]]. In a previous study, we demonstrated that 1, a septanose analog of the FimH ligand n-heptyl α-d-mannopyranoside (2) (Fig. 1), is able to bind to the mannose binding site [22]. The respective crystal structure revealed that the septanose ring can assume a conformation superimposable with the mannose hydrogen bond network in the FimH binding site (Fig. 2A). However, the conformational flexibility of the septanose ring in solution ultimately imposed an energetic burden and resulted in an entropic penalty reducing the overall affinity by a factor of ten.
With this insight into the consequence of exchanging the carbohydrate scaffold itself, the present study aims to leverage the increased flexibility of septanoses to access additional exit vectors for aromatic moieties targeting the so-called tyrosine gate of FimH [23,24]. In contrast to the glycosidic bond in mannosides, which has a strong preference for the gauche-trans or gauche-gauche conformation attributed to the exo-anomeric effect [25], the torsional energy barriers are typically much lower in alkyl ether bonds. Initial modeling studies (see below) indicated that both biphenyl, as well as elongated biphenylmethyl pseudoaglycones were able to adopt a conformation that orients the aromatic moieties towards the tyrosine gate. Thus, both pseudoaglycone series can be seen as functional analogs of biphenyl mannosides, despite their differing chemical structure. FimH is known to undergo a conformational transition from an open state, in which the mannose binding site is solvent accessible, to a closed state with a solvent-shielded mannose hydrogen bond network [19,[26], [27], [28]]. Experimentally, binding to both states can be approximated by employing either the full length protein (FimHFL), in which this mechanism is left intact, or the isolated lectin domain (FimHLD) construct, which is locked in the solvent-shielded conformation unable of undergoing a major conformational reorganization of its binding site. In general, binding to FimHFL is associated with an approximately 100-fold loss in binding affinity due to an entropic penalty associated with the loss of protein flexibility. We hypothesized that more flexible carbohydrate analogs bearing biphenyl modifications, which generated highly potent pyranose ligands for FimH [18,19,21,[29], [30], [31]], might be able to address intermediate conformations along the conformational transition trajectory, altering the associated potential energy landscape, and, thus, enable a more potent interaction with FimHFL.

留言 (0)