
記住我
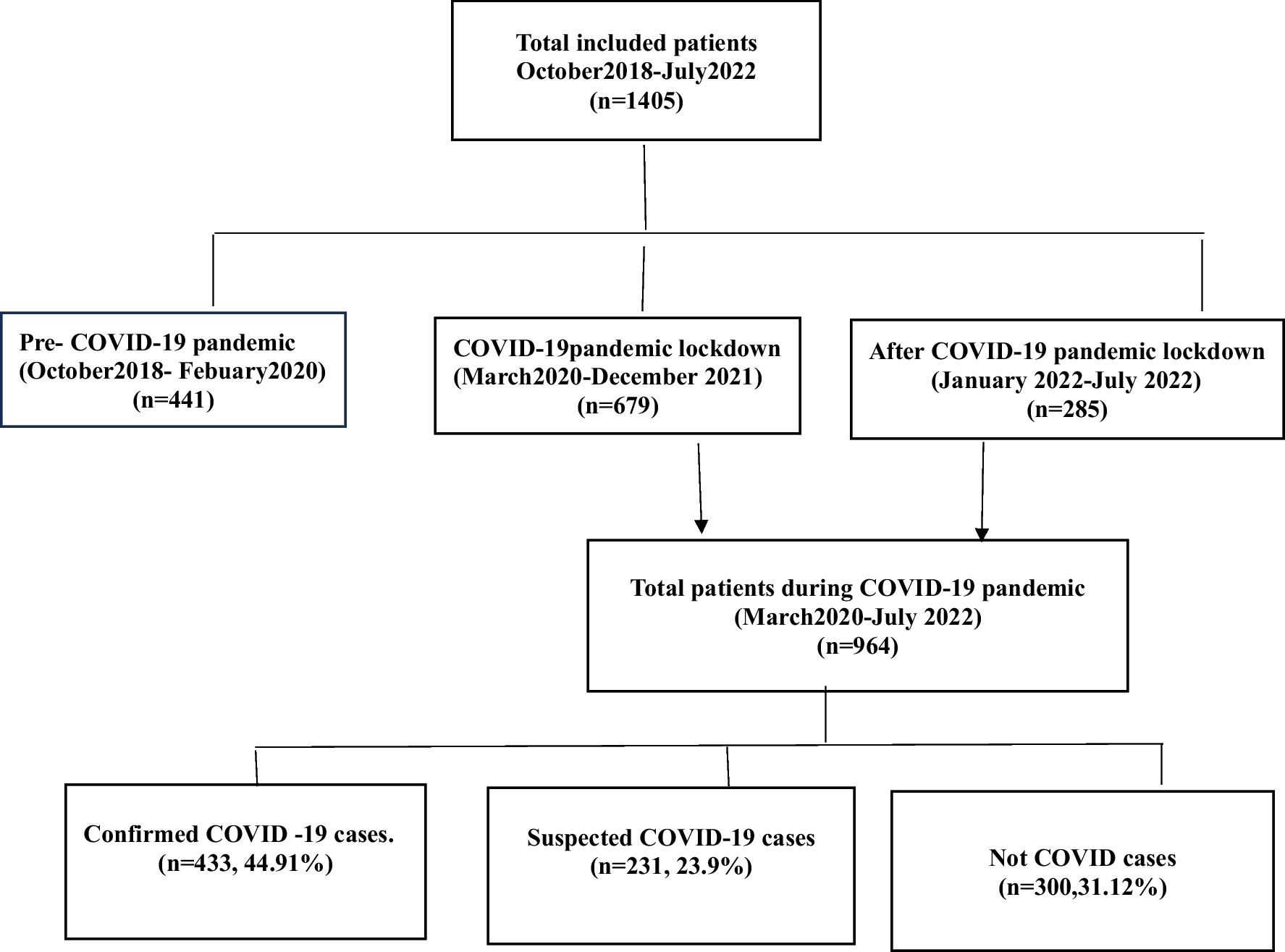





A 46-year-old lady, born to non-consanguineous parentage presented with imbalance while walking since the age of 39 years, progressive, initially was able to walk independently, but gradually needed support over the next 4 years. At age 43, she developed 2 episodes of seizures, semiology suggestive left focal, tonic clonic seizures, however, did not seek medical care for the following. These seizures were mild and not preceded by fever. A year later (at age 44), she was admitted for multiple such episodes involving left upper and lower limb following a febrile episode, leading to epilepsia partialis continua, during which an EEG was done which showed only diffuse slowing of background activity in theta range. The CT brain was normal; however, she declined an MRI brain at that time. She was discharged on a single anti-seizure medication (ASM), Levetiracetam (1000 mg/day), but continued to have intermittent left focal seizures for 2 years and her ataxia worsened. Her family members also noticed cognitive decline and she started becoming dependant for her activities of daily living. At age 46 (2 years later), she presented with multiple episodes of seizure, following a brief episode of an upper respiratory tract infection. Her anti-seizure medications were rapidly escalated to the administration of optimal doses of levetiracetam, Phenytoin, Lacosamide and Clobazam. On clinical examination, she was drowsy but arousable and had decreased movements on her left side. An MRI brain( Philips Achieva,1.5 T, 2011 Model, Country of Origin-Netherlands) showed T2/FLAIR hyperintensities in the right frontal, parietal, occipital cortices, pons, bilateral cerebellar hemispheres, middle cerebellar peduncle, dentate and peridentate region, which were iso-hypointense on T1, hyperintense on DWI (without any drop in ADC map, suggesting shine through phenomenon), without any T1 contrast enhancement (see Fig. 1, 2, 3, 4).
Fig. 1
a DWI image showing hyperintensity involving right middle frontal gyrus and right parietal parasaggital gyrus, b T2 FLAIR image showing hyperintensity in the right middle frontal gyrus and parietal parasaggital region, c T1 image showing corresponding iso-hypointensity, d T2 image showing corresponding hyperintensity
Fig. 2
T2 FLAIR image showing hyperintensity involving bilateral (L > R) middle cerebellar peduncle, dentate and peridentate region
Fig. 3
a DWI image showing transverse hyperintensity involving mid-pons, b T2 FLAIR image showing corresponding hyperintensity, c T1 image showing corresponding iso-hypointensity, d T2 image showing corresponding hyperintensity
Fig. 4
EEG in reference montage showing focal epileptiform discharges from right posterior head region (T4, T6, O2) with background slowing
The above symptom complex and radiological findings posed a diagnostic dilemma. Immune-mediated inflammatory diseases of the nervous system were considered as a possibility; however, lumbar puncture done revealed a normal CSF picture and an autoimmune encephalitis panel (NMDA, VGKC, GABA A/B, MOG Ab) was also negative. Possibility of a mitochondrial cytopathy was thought of; however, extensive literature review failed to reveal the imaging feature [2] of a transverse pontine signal change in this clinical context, further perplexing us. An EEG was performed and showed the findings described in the image. Since all other diagnostic options were exhausted, genetic analysis was sent, awaiting which patient was managed with optimal doses and addition of ASMs, a single pulse of steroids and supportive measure. Genetic testing revealed homozygous missense variation in exon 13 of the POLG gene on chromosome 15. She was initiated on a mitochondrial cocktail and maximal supportive measures, despite which she succumbed to the illness after 2 months.
Our patient’s presentation of ataxia, cognitive impairment with adult onset epilepsy initially led us to believe the possible differentials of a treatable cause like autoimmune encephalitis, however, after the autoimmune encephalitis panel reported negative, and considering prolonged history of illness, we investigated for less common possibilities like a mitochondrial cytopathy [3].
Primary mitochondrial disorders (PMDs) are a relatively rare, clinically variable group of diseases that occur secondary to pathological alterations in mitochondrial DNA (mtDNA) or nuclear DNA (nDNA leading to a multisystem involvement. Among these POLG mutations were identified to be an incriminating factor in 2001[4]. Following this, several clinical phenotypes have been identified [5, 6].
Over 80% of patients affected by POLG-related epilepsy harboured at least one of the three common pathogenic variants, which are p.Ala467Thr, p.Trp748Ser, and p.Gly848Ser [1]. Our patient had a homozygous missense variation in exon 13 of the POLG gene (chr15:g.89323426C > G) which resulted in the amino acid substitution of Serine for Tryptophan at codon 748 (p.Trp748Ser).
Our case highlights the variable nature of the presentation of POLG gene mutation, the difficulty to diagnose it due to the clinical overlap of syndromes and the challenges of the management of mitochondrial epilepsy [7]. We propose that POLG-related mitochondrial disease should be a differential diagnosis in cases of de novo status epilepticus, particularly with other clinical features, such as ataxia [7] and cognitive impairment, as epilepsy is a poor prognostic factor in POLG mutations and late-onset epileptic encephalopathy is uncommon. [7] The clinical and radiological phenotype among patients with a POLG mutation is highly variable. The most commonly encountered findings are areas of diffusion restriction and T2 Flair hyperintensities, with a predilection for the occipital lobes followed by thalamic, basal ganglia and cerebellar lesions, generalised brain atrophy and cerebral white matter [8] signal changes in decreasing order of occurrence [9]. However, in our patient in addition to the cortical, thalamic and cerebellar signal changes, there was a distinct transverse pontine signal change which was iso to hypointense on T1, hyperintense on T2 and Flair with hyperintensity noted in diffusion images, without ADC drop and no contrast enhancement. This is a unique finding, not previously described in a patient with genetically proven POLG mutation. Diffusion restriction and focal neurological deficit in the context of a mitochondrial disease is a characteristic features of mitochondrial myopathy encephalopathy lactic acidosis and stroke like episodes (MELAS) syndrome. Similar to MELAS and in keeping with the molecular basis behind the diffusion restriction being non ischemic in nature, our ADC maps revealed no drop.
The management in patients who carry POLG variants is extremely challenging, and often futile in those presented with recurrent status epilepticus and progressive encephalopathy [1]. Refractory epilepsy is a poor prognostic factor and often associated with mortality.
Unfortunately, there are no successful established treatment modalities except for supportive care and avoidance of hepatotoxic medications due to the predisposition for liver dysfunction [10, 11]. We hence avoided valproic acid in the list of medications prescribed and titrated other ASM s and initiated her on a cocktail of medications to attempt and reduce the oxidative stress.
Peculiarities of this case included the age of presentation, unique combination of clinical features and novel radiological association. While the median age of presentation in a mitochondrial cytopathy is highly variable, the more severe forms present in childhood or early adulthood. Our patient had a late disease onset with rapid progression and progressed to be lethal within 7 years.
Among the clinical phenotypes recognised in a case with POLG mutation, this patient had an overlap symptomology which did not fit into a defined entity. Furthermore radiological findings in POLG-related diseases reveal a predilection for grey matter [12] and only a single documented case of predominantly white matter pathology exists [8]. These areas are likely involved because of the increased presence of mitochondrial oxidative phosphorylation system (OXPHOS) in these locations. However, pontine involvement is a phenomenon that is a novel finding, which requires more analysis. Our case joins a list of growing case reports expanding the spectrum of POLG-associated diseases.
Unlike other genetically mediated diseases, mitochondrial disease pose a peculiar diagnostic dilemma. Due to the variable expressivity of mitochondria in various tissues, the concepts of homoplasmy and heteroplasmy, disease evolution over time, abnormal mitochondria-related death pathways, the unique characteristics of mitochondrial inheritance and precipitation of disease by external triggers, there are no uniform diagnostic criteria. In this context, establishing a radiological pattern recognition is of paramount importance to guide the clinician to decide on ordering genetic tests especially in a resource limited setting.
留言 (0)