
記住我
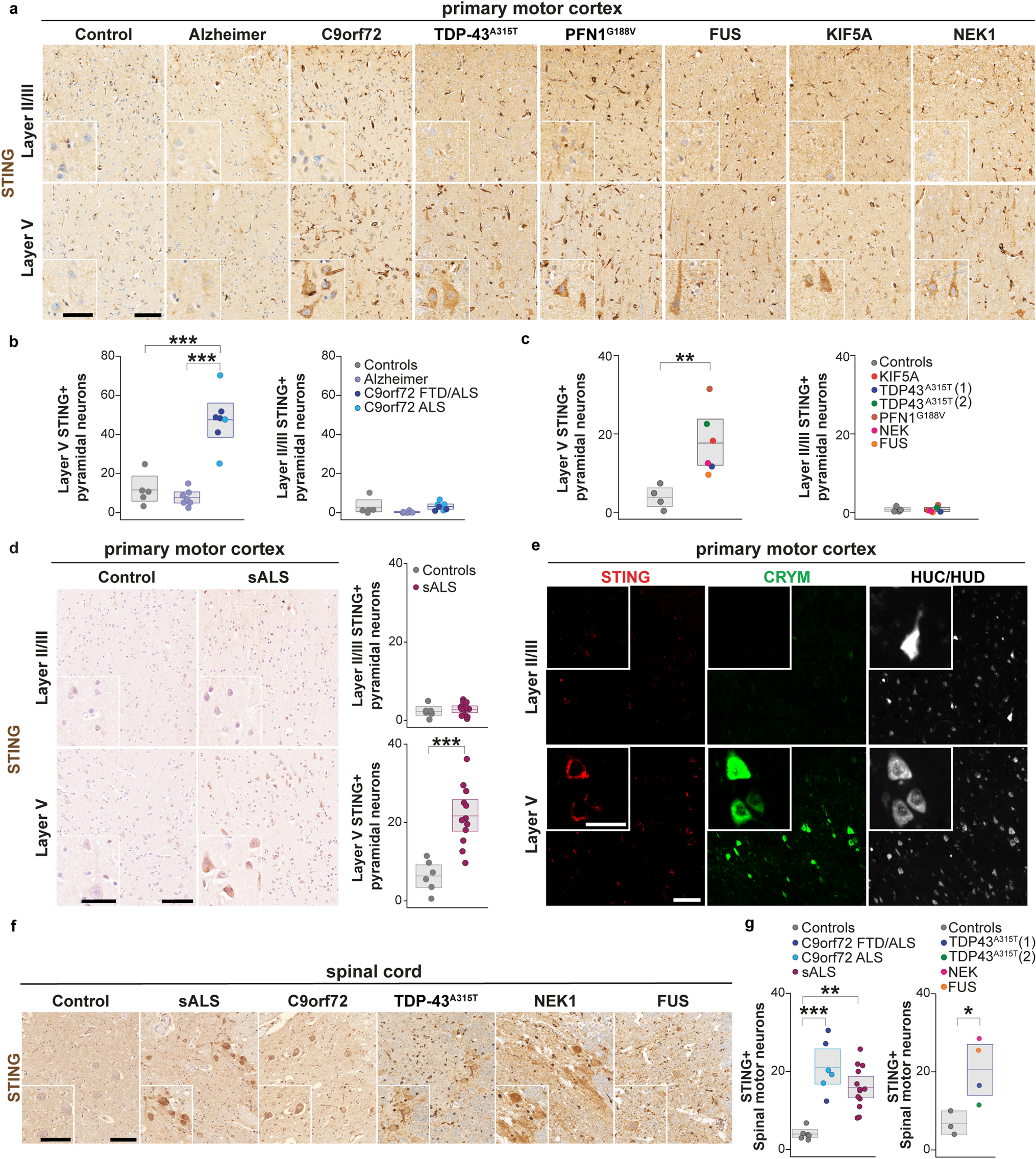

We analysed a study cohort of H3K27-altered gliomas harbouring mutations in BRAF (n = 22), FGFR (n = 37), or both (n = 1; Supplementary Fig. 1 and Table 1, online resource). Of these, 43 were children (< 18 years) and 16 adults. In parallel, we analysed control cases of patients with BRAFWT/FGFRWT DMG H3-K27 from the corresponding cohorts. All DMG H3-K27M with BRAF alterations harboured a somatic V600E substitution, but no fusion (Fig. 1). Concerning FGFR, all cases displayed FGFR1 hotspot substitutions N546K/D (73%, 27/37) and/or K656E/M (23%, 11/37) preferentially occurring in CNS tumours, but neither FGFR1 fusion/duplication nor FGFR2/3 alterations (Fig. 1) [20]. All these mutations in BRAF and FGFR1 are widely known to induce an aberrant MAPK activation [12, 20, 27, 38, 43]. We estimated the overall prevalence of DMG BRAFV600E and DMG FGFR1MUT within the H3.3-K27M tumours in our composite paediatric–adult cohort at 7.6% and 12.3%, respectively. Thus, DMG BRAFMUT/FGFR1MUT together could represent close to 20% of all DMG H3.3-K27M.
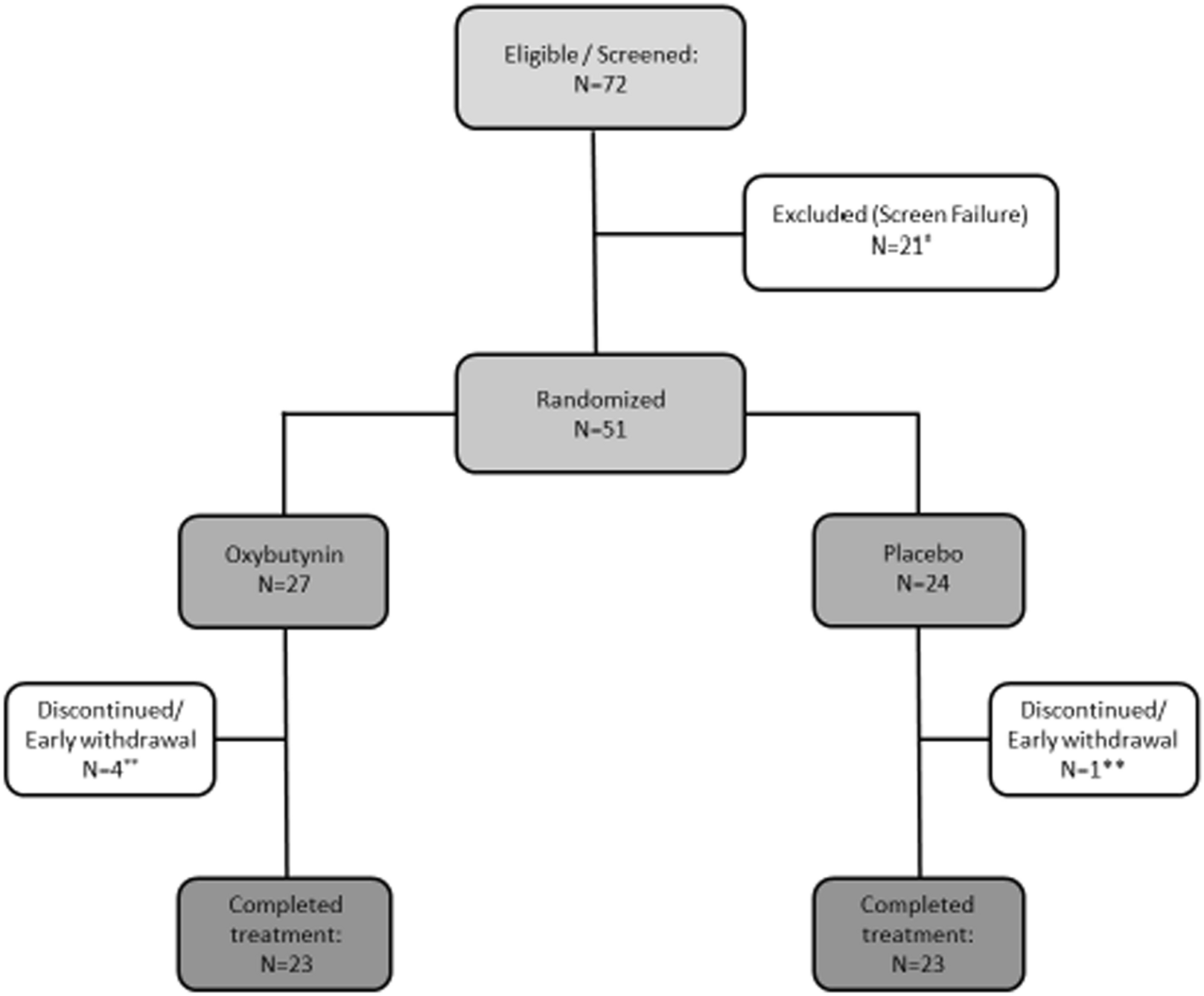
Table 1 Multivariable Cox proportional hazards regression model for the OS of patients with H3-K27M DMGFig. 1
Clinical and molecular characteristics of the patient cohort of DMG H3-K27M with BRAF and FGFR1 mutations. Overview of the clinical and molecular annotations of 60 paediatric and adult DMG H3-K27 patients presenting BRAF or FGFR1 mutations. Cases are presented in columns and genes status in rows. Age is reported in years. All molecular information is derived from DNA or RNA sequencing analyses, except immuno-histological data (including histological profile, EZHIP and ATRX expression). For survival, patients still alive at last follow-up are indicated by a half-filled square
We further analysed the genomic landscape of tumour for which material or data were available and observed that BRAFMUT or FGFR1MUT was mostly associated with H3.3-K27M mutation but not H3.1-K27M mutation. One tumour harboured a FGFR1MUT in the context of a DMG H3-K27 wild-type with EZHIP overexpression presenting an ACVR1 mutation (case #60). Second, BRAF and FGFR1 mutations were mutually exclusive, as only one tumour presented both hits (case #23; Fig. 1). However, no clonality information was available for this case to confirm subclonality of the two mutations. Finally, we observed that in 90% (9/10) of FGFR1MUT cases for which we had the information, FGFR1 mutation was clonal to H3-3A mutation with a similar variant allele frequency suggesting a role in the early steps of oncogenesis of these tumours. It was less frequently the case for patients with DMG H3-K27M BRAFV600E with only 33% (2/6) tumours where BRAF mutation appeared clonal to H3-K27M mutation. In addition, H3-K27 FGFR1MUT tumours presented often other hits in the MAPK pathway with NF1 (13/31; 42%) or PTPN11 in (3/22; 13.6%) as the topmost mutated genes (Fig. 1). Additional MAPK-activating mutations seemed to be less frequent for BRAFMUT tumours with 1/12 (8.3%) case harbouring an NF1 mutation. TP53 mutations were found in 5% (1/20) and 9.3% (3/32) of the BRAFMUT and FGFR1MUT tumours, respectively but not PPM1D (Fig. 1). For one H3.3-K27M FGFR1MUT patient, TP53 mutation was clonal to H3.3-K27M and it was sub-clonal for the other case for which information was available. In addition, ATRX was mutated, or its expression was lost on IHC in 59.3% (16/27) of FGFR1MUT DMG, albeit never in BRAFMUT tumours, and this was not associated to TP53MUT in all but one case (#24). A mutation in a member of the PI3K/AKT/mTOR signalling pathway was present in 19% (4/21) of H3-K27M-FGFR1MUT tumours (Fig. 1).
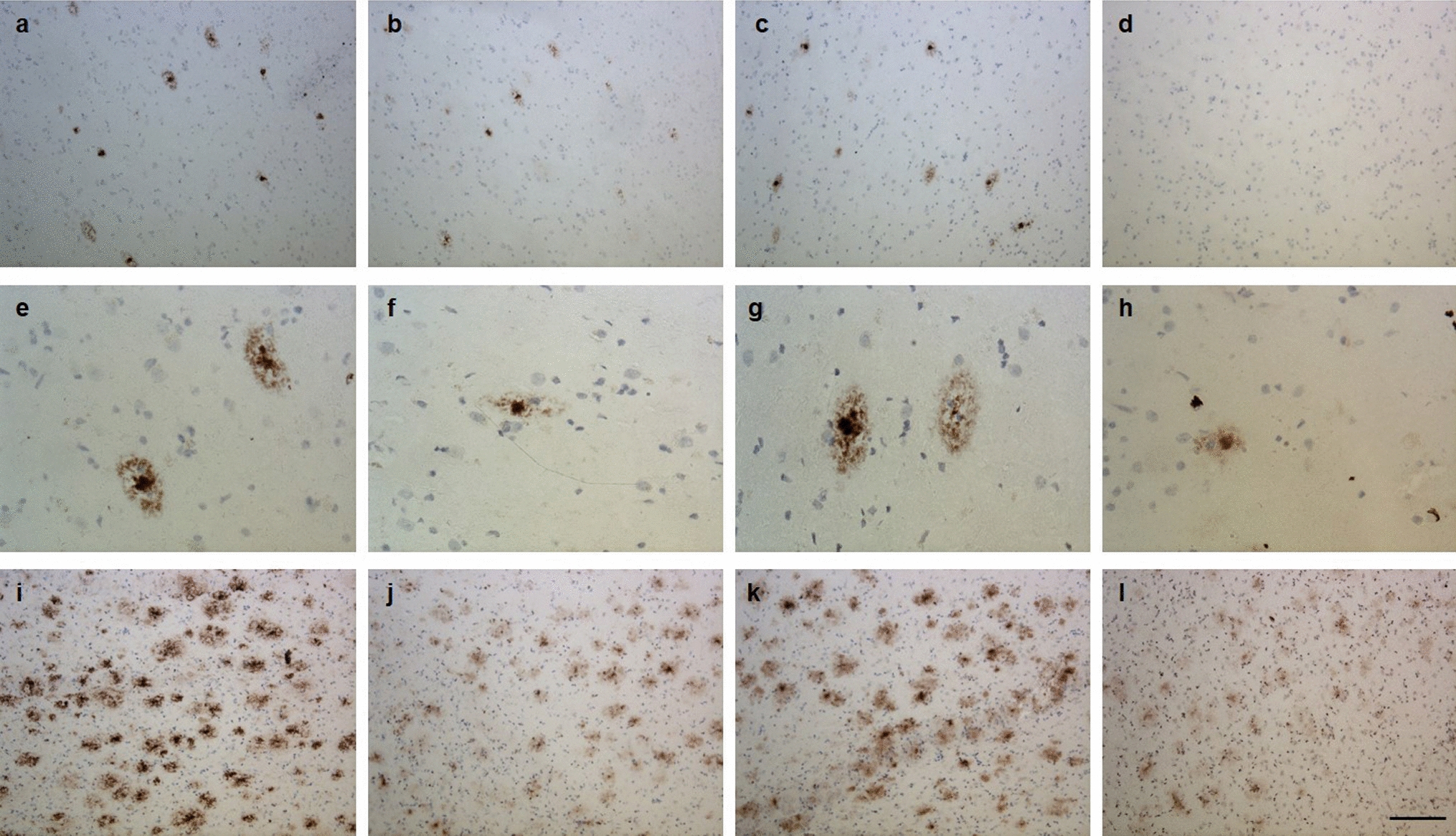
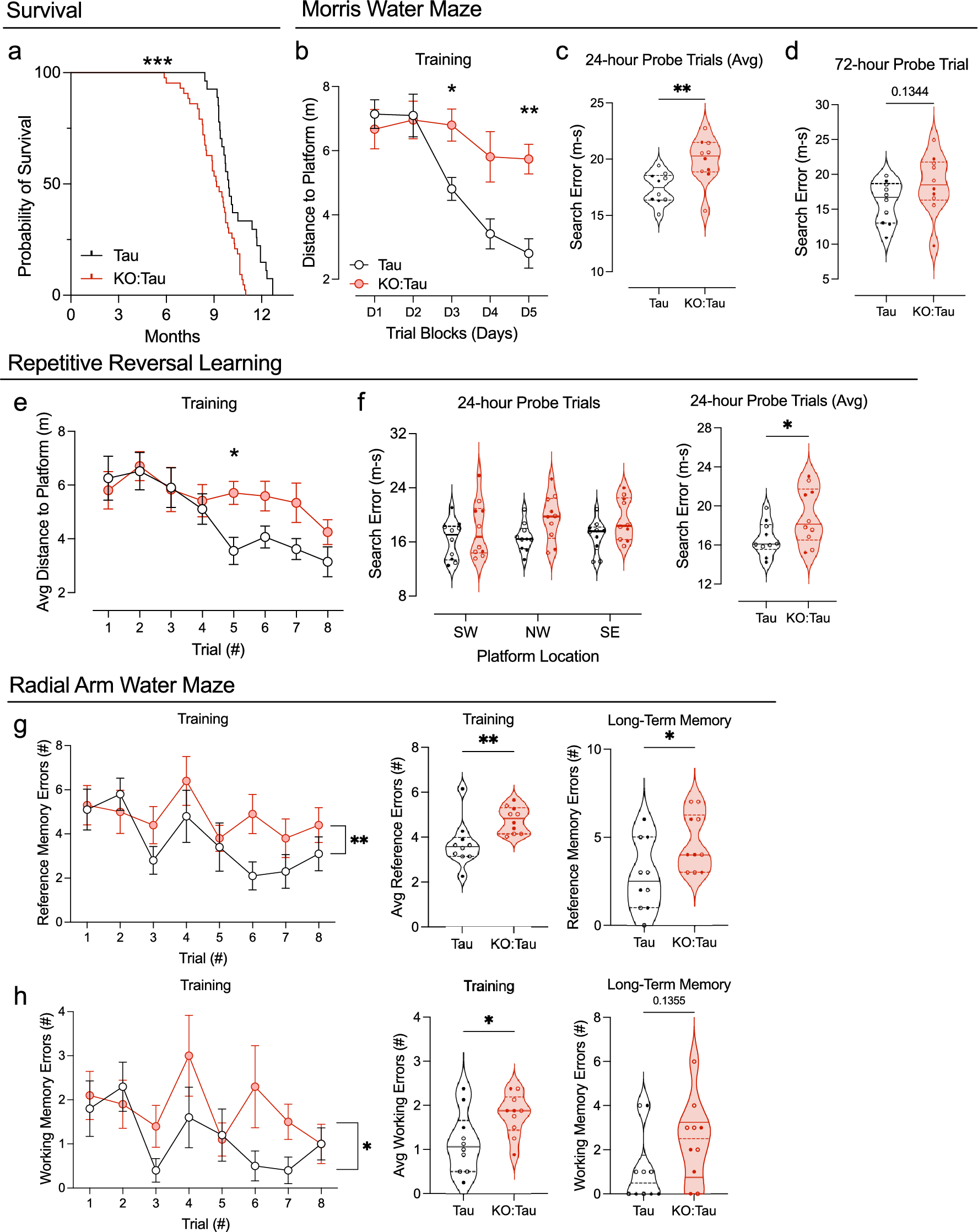
BRAF MUT/FGFR1 MUT H3 K27-altered tumours are histologically heterogeneous with frequent mixed diffuse-circumscribed presentation and calcificationsWe next analysed the histo-pathological profile of cases of our own cohort (Supplementary Table 1). These 29 tumours were initially diagnosed as DMG H3K27 (n = 13), ganglioglioma grade 1 (n = 5), anaplastic ganglioglioma grade 3 (n = 4) or other gliomas (GBM n = 3; pilocytic astrocytoma n = 1; LGG n = 1 or oligo-astrocytoma grade 3 n = 2). All BRAFMUT/FGFR1MUT DMG presented a H3K27 trimethylation loss as expected and were positive for H3K27M staining with the exception of the case #60, harbouring EZHIP overexpression. Tumour growth pattern evaluated morphologically and based on NF70 immunostaining revealed that 12 tumours were only diffuse (n = 3 in FGFR1MUT group and n = 9 in BRAFMUT), 4 were only composed of compact tumoral tissue and 13 were mixed diffuse with nodular compact areas. We observed three major histological patterns: GG-like in 41.3% (12/29), DMG-like in 34.4% (10/29), and HGG with a piloid astrocytic component (piloid-HGG) in 24.3% (7/29). BRAFMUT and FGFR1MUT tumours presented a classic DMG histological profile only in 25% (4/16) and 46% (6/13) of cases, respectively (Figs. 1, 2, Supplementary Fig. 2a, online resource). A GG-like profile, characterised by mixed neuronal and glial tumour cells, was present in 50% (8/16) and 30% (4/13) of BRAFMUT or FGFR1MUT cases, respectively (Fig. 2b–i). Finally, 25% (4/16) of BRAFMUT and 23% (3/13) of FGFR1MUT DMG presented a piloid-HGG profile with piloid cell morphology (Fig. 2j, k). The remaining FGFR1MUT DMG case (3/13) harboured a mixed DMG-like and GG-like or piloid-HGG, and the last GG-like and piloid-HGG profile (Fig. 2l). Micro-calcifications, albeit rare in classic DMG, were observed in 56% (9/16) of BRAFMUT and 38% (5/13) FGFR1MUT DMG (Fig. 2j). Ganglion cells and eosinophilic granular bodies were seen in 37% (11/29) and 31% (9/29) mainly in the BRAFMUT group (7/16) (Fig. 2b). GFAP whorls defined as a small gliofilament tangle, represented a rare but particular aspect (n = 4) (Fig. 2m–o). Perivascular lymphoid infiltrates were frequent (48%, 14/29) and especially enriched in the BRAFMUT group (62%, 10/16) (Fig. 2c). CD34 extravascular staining was observed in the majority of tested cases: 50% (4/8) in FGFR1MUT and 60% (9/15) in BRAFMUT. ATRX loss of expression was frequently seen in FGFR1MUT cases tested (8/11) but absent in BRAFMUT (0/16) (Figs. 1, 2l). The histopathological profile of BRAFMUT or FGFR1MUT DMG thus appeared very heterogeneous and distinct compared to classical DMG H3-K27M.
Fig. 2
Multiple histopathological profiles of DMG H3-K27 with BRAF or FGFR1 mutations. Case 14 a A glioneuronal proliferation with ganglion cells, eosinophilic granular bodies and some microcalcifications (HPS, magnification × 400). Case 11 b A glioneuronal proliferation with numerous ganglion cells (HPS, magnification × 400). Case 13 c A glioneuronal proliferation with numerous ganglion cells and lymphocytic infiltrates (HPS, magnification × 400). Case 31 d A mainly circumscribed proliferation (neurofilament, magnification × 30). Case 32 e A mainly circumscribed proliferation (neurofilament, magnification × 400) with a diffuse component at the periphery of the tumour f (neurofilament, magnification × 400). g Diffuse chromogranin A immunoreactivity staining neuron cells (magnification × 400). h BRAFV600E expression in all tumour cells including ganglion cells (magnification × 400). i H3K27M expression in all tumour cells including ganglion cells (magnification × 400). Case 7 j A glial proliferation with oligo-like features and microcalcifications (magnification × 400). Case 11 k Global loss of H3K27me3 (magnification × 400). (l) Loss of ATRX in tumour cells (magnification × 400). m Whorls of gliofibrillary processes (HPS, magnification × 20). n Whorls of gliofibrillary processes (HPS, magnification × 400), stained using GFAP antibody o, magnification × 400). Black scale bars represent 50 μm (a–c, e–l and n–o), 100 µm (m), and 500 µm (d)
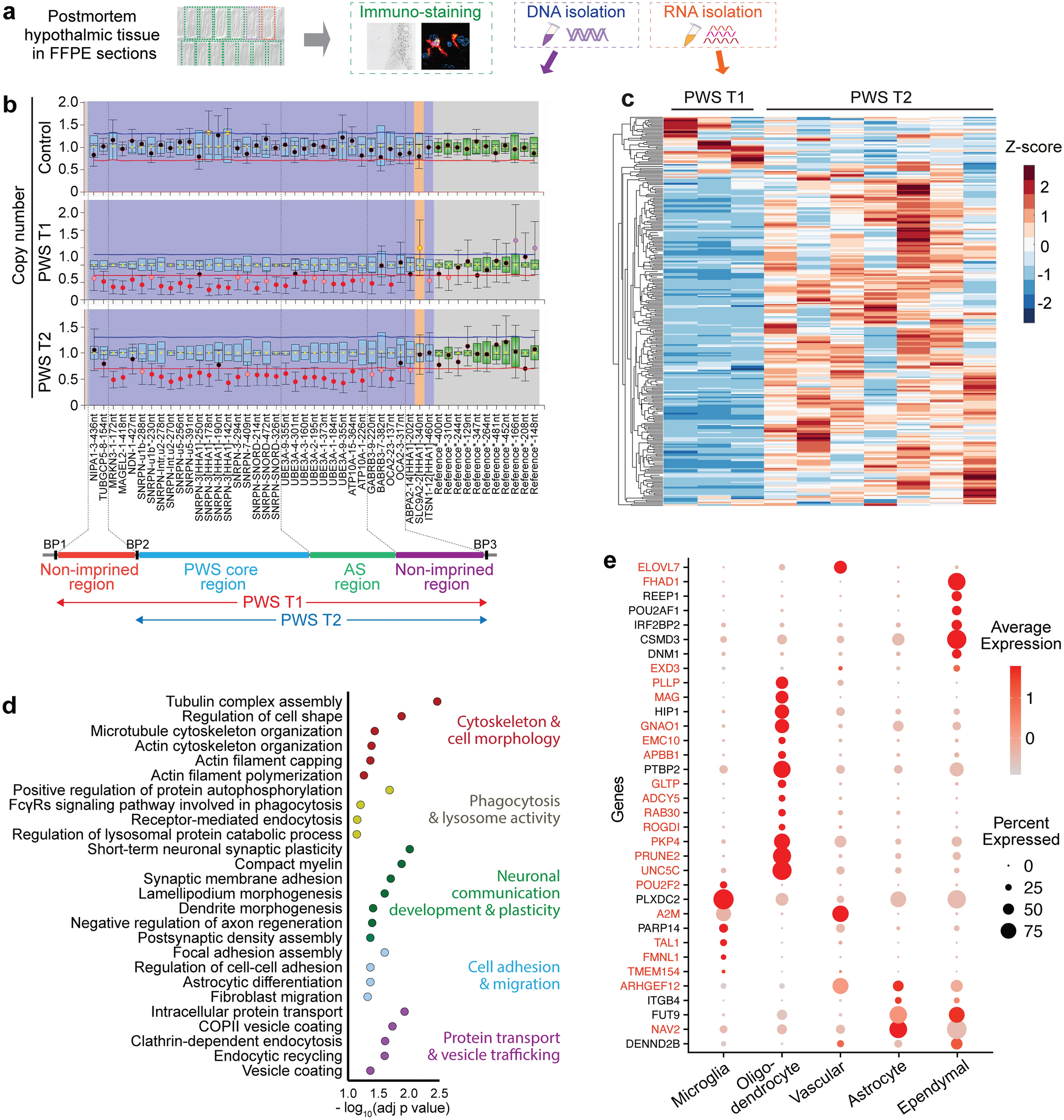
Radiologically, we observed that DMG H3-K27 BRAFMUT or FGFR1MUT were mostly diffuse but presented a significant enrichment of a mixed nodular-diffuse aspect compared to control DMG H3-K27 (Fig. 3a–b; (9/11) 82% and (5/9) 56% versus (4/47) 8%; p value = < 0.0001, chi-square test for trend). Few tumours were completely circumscribed (Fig. 3a, b—(2/11) 18% and (2/9) 22% versus (0/47) 0%; p value = < 0.0001, Chi-square test for trend), and radiological analysis at progression identified a diffuse evolution pattern in half (2/4). Second, these tumours were more often contrast enhancing (Supplementary Fig. 2b, online resource; (11/11) 100% and (9/9) 100% versus 47% (22/47); p value = 0.0013 and 0.0029, respectively; Fisher’s exact test). Third, they developed more calcifications than DMG H3-K27 (Fig. 3a, c; (6/11) 55% and (5/9) 56% versus (1/38) 3%; p value = < 0.0001, Fisher’s exact test). Presence of micro-calcifications detected in histological analyses correlated with macro-calcifications seen by CT scan in 4/8 (50%) DMG BRAFMUT and 2/5 (33.3%) FGFR1MUT (Supplementary Fig. 3c, d, online resource). In total in histologic and radiologic analyses, DMG H3K27M BRAFMUT or FGFR1MUT was calcified in 8/11 (72.7%) and 6/9 (66.7%), respectively. To evaluate how radiological features could detect DMG with MAPK alterations, we classified tumours according to a ‘classic’ (diffuse) or ‘atypical’ (nodular-diffuse or circumscribed aspect and/or presence of macro-calcifications) radiological profile independently of their genotype. This showed that 91% (10/11) of DMG with BRAFV600E and 78% (7/9) DMG with FGFR1MUT H3K27M DMG are classified as atypical versus only 8.5% (4/47) classical DMG H3K27 (Supplementary Fig. 3e, online resource; p value = < 0.0001, Fisher’s exact test).
Fig. 3
Radiological specificities of DMG H3-K27M BRAFV600E or FGFR1MUT. a T2-FLAIR (Fluid-attenuated inversion recovery) MR images sequences or CT scans (computed tomography) of DMG H3-K27M according to BRAF or FGFR1 mutation status. b Comparison of the tumour radiological presentation (diffuse, circumscribed, or mixed) of DMG according to their genotype (chi-square test for trend: ns, ****p value < 0.0001). c Comparison of presence of macro-calcifications in DMG CT-scans according to the presence of MAPK alteration (Fisher’s exact test: ****p value < 0.0001, *p = 0.0210, *p = 0.0348)
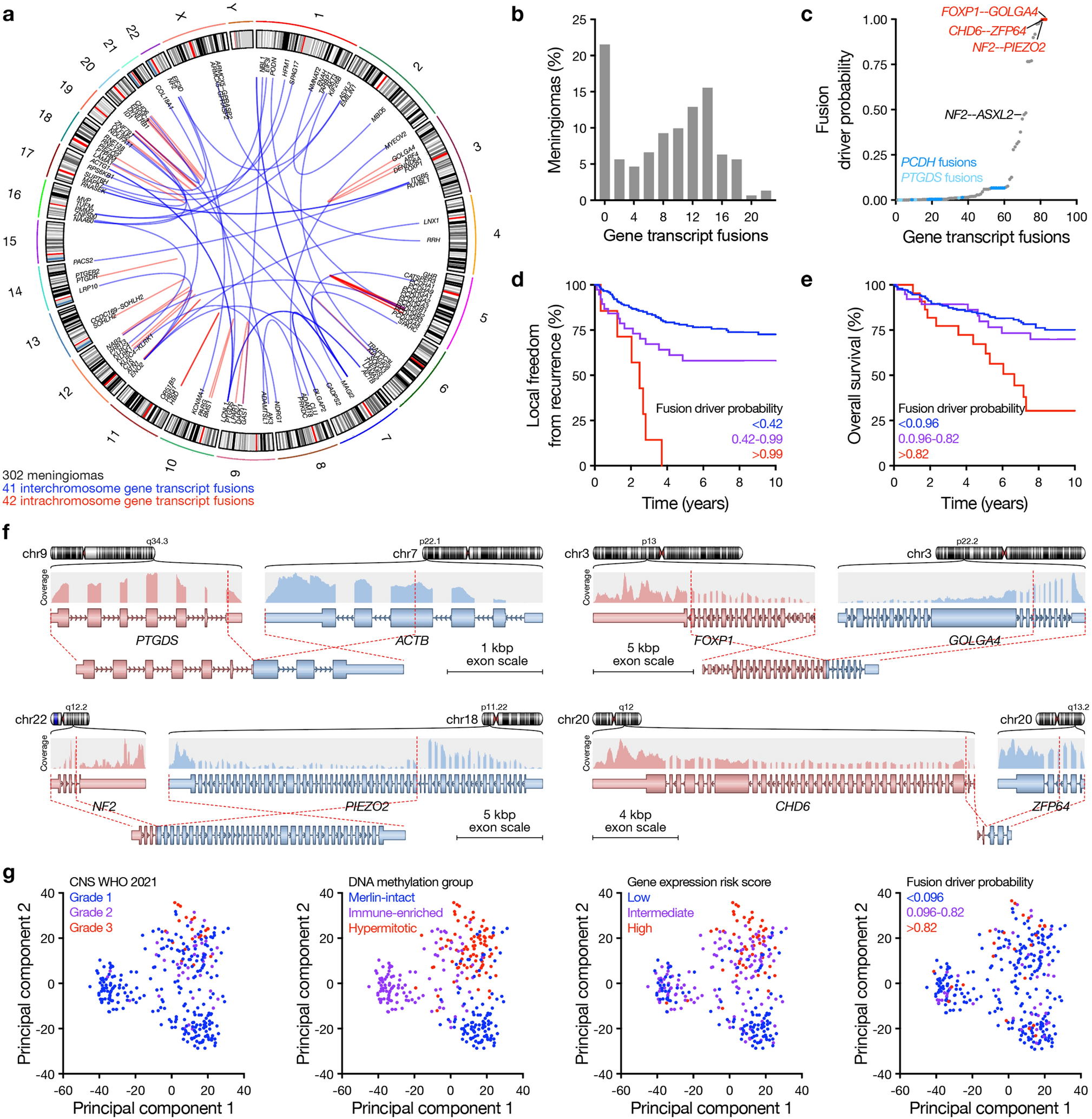
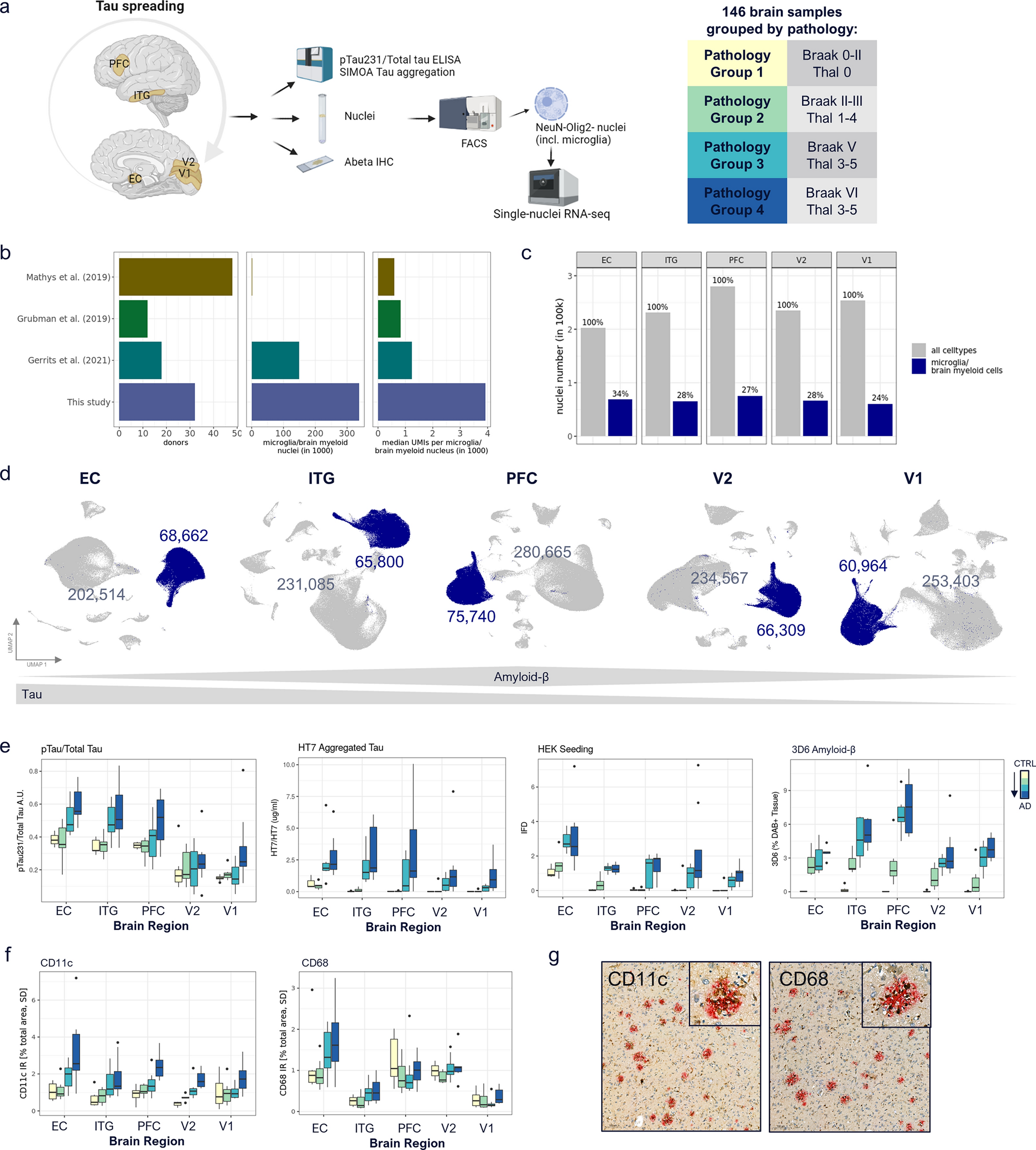
DNA methylation profiling distinguishes a subgroup of DMG H3 K27-altered with MAPK-activating mutationsGiven the disparities between DMG_K27-BRAF/FGFR1 and classical DMG H3K27-altered, we hypothesised that DMG H3 K27-altered with MAPK-activating mutations might correspond to either (i) a new subtype of DMG or (ii) atypical aggressive MAPK-driven low-grade gliomas/glioneuronal tumours. To test these hypotheses, we analysed the DNA methylation profile of the whole cohort. Based on the Heidelberg DNA methylation brain classifier V12.8, 54% (7/13) of BRAFMUT and 67% (10/15) of FGFRMUT tumours classified as DMG_K27 and the remaining corresponded to other classes or were undefined (score < 0.9) (Supplementary Fig. 3a, online resource), which was consistent with the hypothesis that DMG H3 K27-altered with MAPK-activating mutation constitute a unique subtype of DMG. In contrast, all DMG H3.3-K27M BRAFWT/FGFRWT clustered as DMG_K27. We then performed an unsupervised clustering based on DNA methylation profiles of these samples together with reference gliomas from the literature [2]. All DMG H3-K27 BRAFMUT or FGFR1MUT but one (case #42) separated from DMG H3-K27 on the tSNE, independently of their H3 mutational status or brain classifier score (Fig. 4, and supplementary Fig. 3b, online resource). They also separated from midline BRAFV600E LGG and GG grade 1 (Supplementary Fig. 3c, online resource). Furthermore, they were grouped in subclasses according to the nature of the secondary MAPK mutation in BRAF or FGFR1 (Fig. 4, and supplementary Fig. 3b, online resource).
Fig. 4
Analysis of DNA methylation profiles of DMG H3.3-K27M BRAFMUT/FGFR1MUT. CNS tumour classification based on DNA methylation profiles. Unsupervised clustering by t-SNE analysis of tumours based on their DNA methylation profiles using 10,000 topmost differentially methylated probes across the reference sample set composed of samples from Capper et al. (n = 936) and Castel et al. (n = 41)
BRAF and FGFR1 mutational status are prognostic in paediatric and adult DMG H3 K27-alteredTP53 mutations and BRAFV600E/FGFR1 mutations are mostly mutually exclusive in DMG. In order to avoid the confounding effect of TP53 mutations on the outcome of H3.3-K27M DMG without MAPK-activating alterations, we stratified patients on Histone H3 and TP53 genotypes of both subgroups in Kaplan–Meier overall survival (OS) analyses. This analysis showed a significant better OS for paediatric and adult DMG H3-K27 patients with activating BRAF (median OS 37 mo.) or FGFR1 mutations (median OS 36 mo.) compared to DMG H3.3-K27M TP53WT (median OS 12 mo.) and other DMG subtypes (Fig. 5a; p value < 0.0001, global log-rank test). Further, we showed that there was no impact of histopathological features such as microvascular proliferation, necrosis or mitotic index, on the OS of patients with BRAF or FGFR1-mutated DMG (supplemental Fig. 2f-h, online resource). To pursue, we performed a multivariable analysis to evaluate the association of Histone H3, BRAF, FGFR1 and TP53 mutational status, age at diagnosis and tumoral location with survival. As expected from previous publications, histone H3 and TP53 status were significantly associated with OS (Table 1) [4, 41]. Age was significantly associated with prognosis, but its overall impact was only marginal compared to other variables. We also showed that BRAFV600E (HR: 0.2132, 95% CI 0.1098–0.4140, p value = 5.03e-06) and FGFR1MUT (0.3414, 95%CI 0.1963–0.5939, p value = 0.0001) are strong and independent prognostic markers in H3K27-altered DMG (Table 1).
Fig. 5
Clinical specificities of patients with DMG H3-K27M BRAFMUT/FGFR1MUT and transcriptomic tumour profiling. a Comparison of OS estimated using Kaplan–Meier method according to the mutation status of Histone H3, BRAF, FGFR1 and TP53 (log-rank test, p value = < 0.0001). b Distribution of age at diagnosis according to BRAF and FGFR1 mutation status (Mann–Whitney test; *p value = 0.0168, ***p value = 0.0001, ****p value = < 0.0001). c Comparison of tumour location according to the mutation status of BRAF and FGFR1 (chi-square test for trend: ns, ***p value = 0.0004 ****p value = < 0.0001). d GSEA plot showing common transcriptomic signature from DMG H3.3-K27M vs. DMG H3.3-K27M BRAFV600E or FGFR1MUT. The normalised enrichment score (NES) and the false discovery rate (FDR q) are indicated in each plot
Clinical disparities in paediatric and adult patients with DMG H3-K27M BRAF/FGFR1-mutatedWe next compared other clinical parameters. The sex ratio was balanced in DMG with MAPK-activating mutations (Supplementary Fig. 4a, online resource). However, we identified a significant difference in age at diagnosis between H3.3-K27M DMG, BRAFMUT and FGFR1MUT DMG (Fig. 5b). More precisely, H3.3-K27M BRAFV600E DMG developed only in children (< 18 years) with a median age at onset of 7.2 years lower than DMG H3.3K27M with 9.85 years (Fig. 5b; p value = 0.0123, Mann–Whitney test) but similar to DMG H3K27-altered paediatric cases with 7.6 years in a restricted paediatric cohort (supplementary Fig. 4b, online resource). In contrast, patients with FGFR1MUT have a significant higher age at diagnosis compared to those FGFR1WT with median of 14.8 and 9.8 years respectively (Fig. 5b; p value = 0.0001, Mann–Whitney test). The age at diagnosis of H3-K27M DMG can vary according to initial tumour locations, with a higher age of onset for thalamic versus pontine tumours [18, 34]. We found this difference of age according to tumour location in DMG H3.3-K27M and in DMG BRAFV600E but not in DMG FGFR1MUT (Supplementary Fig. 4c, online resource). Finally, DMG with BRAF and FGFR1 mutations were significantly more frequent in the thalamus compared to DMG H3.3-K27M (Fig. 5c; 70% (16/23) and 58% (21/36), respectively versus 28% (55/199); p value = 0.0004, and p value < 0.0001, Chi-square test for trend). No variation in term of OS, age at diagnosis and tumour location was noted according to FGFR1-mutated variant: FGFR1N546K/D versus FGFR1N656E (Supplementary Fig. 5, online resource).
H3.3-K27M mutation occurs prior to BRAF V600E during oncogenesisWe next wondered if the sequence of appearance of mutations was identical in these tumours, and more largely if they correspond to (i) DMG (H3-K27M as a first hit) or (ii) atypical aggressive low-grade gliomas (BRAFV600E as first hit). To address this question, we analysed BRAF copy number variation (CNV) by digital droplet Polymerase Chain Reaction (ddPCR) on genomic DNA from one BRAFV600E H3.3-K27M tumour (case #2) and from a H3.3-K27M BRAFWT clone derived from this primary tumour in vitro (Supplementary Fig. 6, online resource). The BRAFWT clone had two BRAF alleles and thus resulted from the re-amplification of an ancestral H3.3-K27M-only clone, but not from a genetic loss of the BRAFV600E allele, demonstrating that, in one DMG H3.3-K27M BRAFV600E, H3.3-K27M was the first hit. Interestingly, DNA methylation profiles from this in vitro amplified BRAFWT H3.3-K27M ancestral clone from case #2 and also from a tumour relapse enriched in H3.3-K27M BRAFWT clone from patient #7 (initially diagnosed with DMG H3.3-K27M BRAFV600E), clustered with DMG H3-K27 with MAPK alterations instead of classical DMG H3-K27 (Supplementary Fig. 3b, online resource).
DMG H3.3K27M with MAPK alterations show a transcriptomic signature of senescence with up-regulation of CDKN1A (P21)In order to investigate the possible specificities of the DMG H3.3-K27M with BRAFMUT or FGFR1MUT, we compared their transcriptome to regular DMG H3.3-K27M TP53WT separately. We found 676 significantly up-regulated and 633 down-regulated genes in the contrast H3.3K27M BRAFWT versus H3.3-K27M BRAFV600E DMG and 228 up-regulated and 274 down-regulated genes in the contrast H3.3-K27M FGFR1WT versus H3.3-K27M FGFR1MUT DMG (adj. p value ≤ 0.001) (Supplementary Fig. 7a, b, online resource). Ninety-four up-regulated and 111 down-regulated genes were common to the two comparisons. Using gene set enrichment analyses (GSEA), we observed an enrichment for MAPK signalling and PI3K/AKT/MTOR signalling signatures in both BRAFMUT and FGFR1MUT DMG (Fig. 5d; Supplementary Fig. 7e, f, online resource) as well as angiogenesis and hypoxia signatures (Supplementary Fig. 7 g-j, online resource). In addition, transcriptomic signatures highlighted activation of senescence and P53 signalling pathway in both comparisons (Fig. 5d). P53 protein is a tumour suppressor implicated in the permanent cell cycle arrest by inducing senescence or apoptosis in response to stress like oncogene activation [6, 14]. TP53 pathway activation was validated at the protein level by immunohistochemistry (IHC) in 71% (10/14) of DMG H3.3-K27M TP53WT with BRAF/FGFR1 alterations, showing heterogeneous, weak to strong TP53 staining. Moreover CDKN1A, encoding the senescence marker P21, was overexpressed at the RNA level in both BRAFMUT and FGFR1MUT gliomas compared to those only H3.3-K27M mutated (adj. p value < 0.0001) (Supplementary Fig. 7 k online resource). CDKN2A, which encodes the tumour suppressor P16, was overexpressed only in BRAF-mutant gliomas (Supplementary Fig. 7 l online resource; adj. p value = 0.0048). As a whole, based on transcriptome and IHC, DMG H3-K27 with BRAFMUT/FGFR1MUT were characterised by a senescence programme, likely induced trough a P16/P21-P53 axis.
留言 (0)