
記住我
The EPIC-Norfolk study is a cohort of 25,639 men and women aged 40–79 years at baseline in 1993–1997, which has previously been described in detail [16]. The EPIC-Norfolk study was approved by the Norfolk Research Ethics Committee (ref. 05/Q0101/191); all participants gave their informed written consent before entering the study.
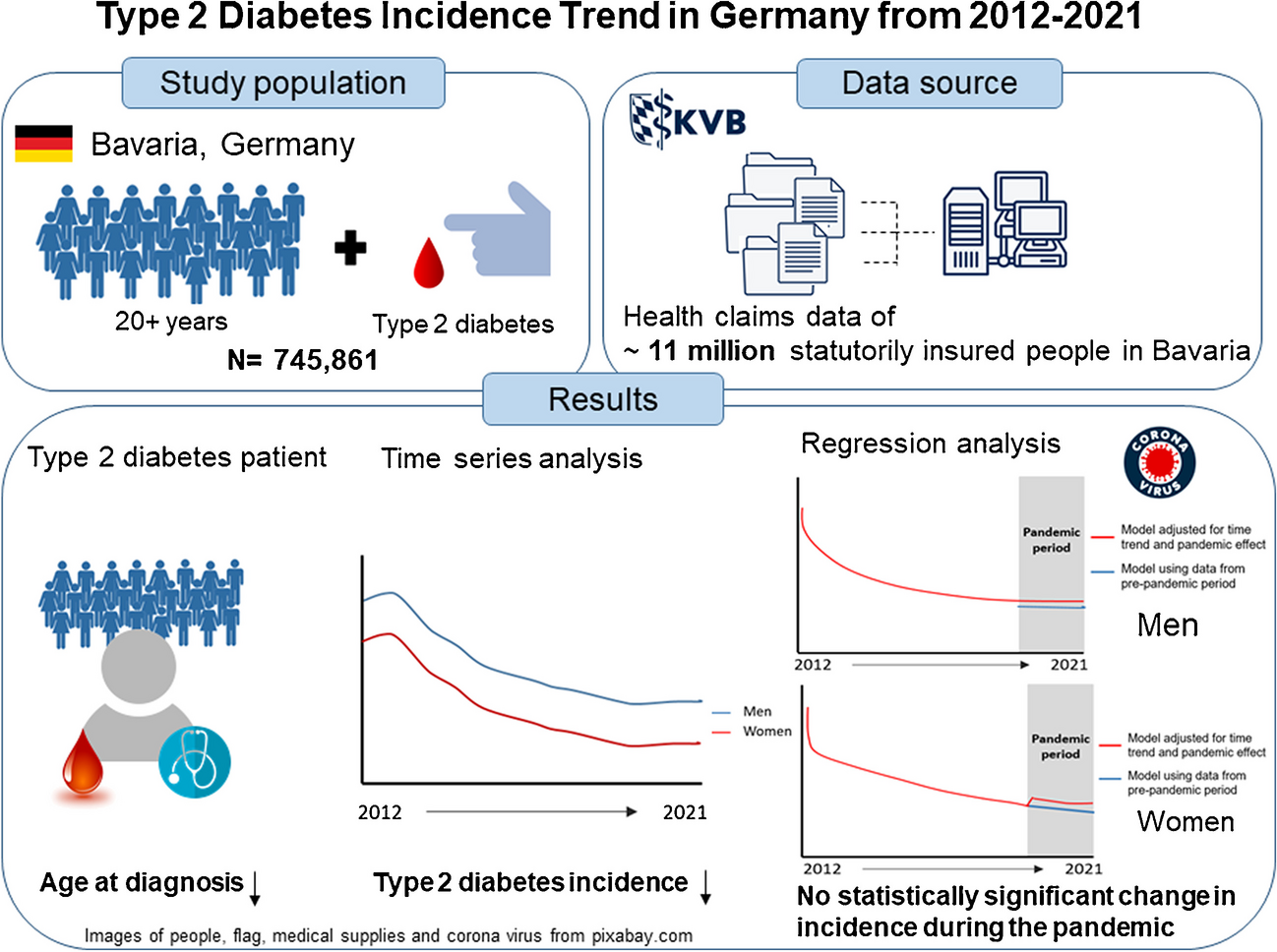
Here, we designed a prospective type 2 diabetes case-cohort (Fig. 1) based on all individuals with no evidence of prevalent diabetes at baseline and available stored blood samples [17]. We ascertained and verified all individuals who developed incident type 2 diabetes over 10 years (follow-up was censored at date of type 2 diabetes diagnosis up to 31 December 2007) based on self-report, doctor-diagnosed diabetes, diabetes drug use or evidence of diabetes after baseline with a date of diagnosis prior to the date of baseline visit, as described previously [18]. We randomly selected 875 individuals from eligible participants as a control cohort. Participants with evidence of prevalent diagnosed diabetes, or those with prevalent diabetes but undiagnosed at baseline, HbA1c ≥48 mmol/mol (6.5%), were excluded. We further used the WHO threshold to define prediabetes according to HbA1c levels as ≥42 mmol/mol (6.0%) and <48 mmol/mol (6.5%).
Fig. 1
Study design. We designed a case-cohort (N=1105) for incident type 2 diabetes within the EPIC-Norfolk study. Genotyping, proteomics (SomaScan v4), metabolomics (Metabolon Discovery HD4) and biomarker profiling were done in samples from the baseline assessment. T2D, type 2 diabetes. Created with BioRender.com
Type 2 diabetes PGSGenome-wide genotyping was performed using the Affymetrix UK Biobank Axiom Array (Thermo Fisher Scientific, Santa Clara, USA), with imputation to the Haplotype Reference Consortium r1.0 and the UK10K plus 1000 Genomes phase 3 reference panels. We generated a genome-wide PGS for type 2 diabetes using LDpred2 [19] (bigsnpr R package v1.8.8 [20]), which has been shown to outperform traditional pruning and thresholding methods. Briefly, this method uses a Bayesian approach that relies on genome-wide summary statistics and linkage disequilibrium (LD) information from an external reference panel to infer posterior mean effect size for each of the variants. Quality control for individual-level genotype data was performed using PLINK v1.9 (https://www.cog-genomics.org/plink/1.9) [21, 22]. We removed strand ambiguous variants with a minor allele frequency (MAF) <1%, Hardy–Weinberg equilibrium p<1×10−6 or a missing rate >1%. Individuals with a genotype missing rate >10% or those with a first- or second-degree relative in the sample were removed (N=17). We further restricted PGS generation to HapMap3 variants as suggested by the LDpred2 authors. PGSs were generated using the infinitesimal model option based on 721,911 variants. Summary statistics were obtained from the largest European meta-analysis including 228,499 type 2 diabetes cases [9].
Omics profilingFor the individuals included in the case-cohort, we used citrate plasma samples that had been stored in liquid nitrogen since the baseline assessment, which took place between 1993 and 1997, for proteomics profiling using the SomaScan v4 assay (SomaLogic, Boulder, USA). Assay details have been described previously [14]. Briefly, this assay uses 4979 aptamer reagents to target 4775 unique proteins. Normalisation was performed by SomaLogic using adaptive normalisation by maximum likelihood (ANML). For all analyses, protein relative fluorescence intensities were log10-transformed and scaled to have a mean of zero and variance of 1.
Untargeted metabolomics profiling was done in samples from the baseline visit for individuals included in the case-cohort using the Metabolon Discovery HD4 platform, as previously described [23] (Metabolon, Durham, USA). We kept data from only 762 metabolites with no more than 50% of missing values overall and no more than 50% of missing values in incident type 2 diabetes cases. Missing values were imputed using the missForest v1.4 R package [24]. Metabolites were natural log-transformed and scaled. We further measured 17 biomarkers and again imputed missing values with the missForest R package. Transferrin, albumin, alkaline phosphatase (ALP), alanine aminotransferase (ALT), apolipoprotein (Apo)A1, ApoB, aspartate aminotransferase (AST), C-reactive protein (CRP), ferritin, GGT, iron and uric acid were measured by standard immunoassays (Olympus AU640 Analyzer). Total cholesterol, triglycerides and HDL-cholesterol were analysed on an RA-1000 (Bayer Diagnostics, Basingstoke, UK). LDL-cholesterol was calculated with the Friedwald formula [25], except for when triglyceride levels were >4 mmol/l. Vitamin C levels were measured with a fluorometric assay from plasma that was stabilised using metaphosphoric acid [26]. HbA1c was measured as part of the EPIC-Interact project using a Tosoh (HLC-723G8) assay on a Tosoh G8 analyser.
Derivation and validation of predictive modelsWe excluded participants with missing genotype data, HbA1c or information on variables included in the Cambridge Diabetes Risk Score [27], leaving 1105 participants (10,792 person-years of follow-up) for analyses (375 incident type 2 diabetes cases) (electronic supplementary material [ESM] Fig. 1a). To have an independent internal validation set, completely blinded to any previous feature selection and hyperparameter tuning steps, we divided individuals into a training set (80%, N=884) and a testing set (20%, N=221). We trained models using regularised Cox regression, including a patient-derived information model, which is based on variables used for the Cambridge Diabetes Risk Score (age, self-reported sex, family history of diabetes, smoking status, prescription of antihypertensive medication and BMI) [27], and a standard clinical model (including the variables from the Cambridge Diabetes Risk Score and HbA1c). We refitted a model using variables from the Cambridge Diabetes Risk Score to find optimal weights in EPIC-Norfolk and to enable a fair comparison among all models.
For omic predictors, we first performed feature selection in the training set. Feature selection was carried out by least absolute shrinkage and selection operator (LASSO) to identify the top predictors among the proteome, the metabolome and 17 clinical biomarkers. A nested tenfold cross-validation (inner loop to determine regularisation parameter, ʎ) was done over 100 subsamples, taking 80% of the training set (outer loop). Within each omic layer, we ranked each feature based on an absolute weighted sum of the number of times it was included in the final model from each of the 100 subsamples and selected the ten features with the highest rankings separately for each omic layer. We additionally performed feature selection across all omic layers (including all proteins and metabolites and the type 2 diabetes PGS) to identify the top ten omic predictors. We implemented this workflow using the R packages caret v6.0-89 [28] and glmnet v4.1 [29]. Features selected were taken forward for parameter optimisation by tenfold cross-validation of the model by regularised Cox regression (incorporating Prentice weights to account for the case-cohort design [30, 31]) in the entire training set. For each of the omic layers, top predictors were optimised alone or on top of the standard clinical model, for which variables were forced to be kept in the models.
Performance of the classification models was evaluated in the internal independent validation set, which was never used for training and optimisation. The prediction models’ discriminatory power was assessed by computing the C index over 1000 bootstrap samples of the test set. We further tested the model’s performance by stratifying the test set into participants with (N=176) and without prediabetes (defined as HbA1c ≥42 mmol/mol [6.0%], N=46). Performance of PGS-only models was tested using simple Cox proportional hazards models (Prentice-weighted), using an analogous bootstrapping framework. To determine whether the addition of omics improved performance over the clinical model, we estimated one-sided p values for the difference in the mean C indices (from bootstrapping). This was calculated as the number of times the difference (between the C indices from clinical + omics and the clinical model) was lower than zero divided by 1000 (that is, the number of bootstrap C indices). For each of the omic layers, we calculated the net reclassification improvement (NRI) when added on top of the clinical model in the test set (all and stratifying by the HbA1c threshold for prediabetes) using the R package nricens v1.6 [32].
Cumulative type 2 diabetes incidenceTo be able to compute absolute risk estimates, we leveraged genotype information available for participants from the entire EPIC-Norfolk study. Linkage with hospitalisation and death registry data was done using UK National Health Service (NHS) numbers through NHS Digital. Vital status was ascertained for the entire EPIC-Norfolk cohort and death certificates were coded by trained nosologists according to ICD-10. For type 2 diabetes definitions, participants were identified as having an event if the corresponding ICD-10 code was registered on the death certificate (as the underlying cause of death or as a contributing factor) or as the cause of hospitalisation. Since the long-term follow-up of EPIC-Norfolk included a period in which coding shifted from ICD-9 to ICD-10, codes were consolidated. We computed the predicted risk for incident type 2 diabetes using the clinical + PGS model in the entire EPIC-Norfolk cohort, excluding individuals with HbA1c levels above the threshold for diabetes (≥48 mmol/mol [6.5%]), with incomplete data for the variables included in this model or who were included in the training set from the case-cohort study (N=9009, ESM Fig. 1b). We stratified participants into those with prediabetes according to their HbA1c levels (≥42 mmol/mol [6.5%]) and quartiles of predicted risk according to the clinical + PGS model among individuals with normoglycaemia. We estimated the cumulative incidence of type 2 diabetes over a 20 year follow-up period in these five strata as 1 minus the Kaplan–Meier estimate of the survivor function.
留言 (0)