
記住我

Two types of cell culture media were used for incubation of splenocytes during experimental procedures. Medium A was RPMI 1640 (Biochrom-Merck, Darmstadt, Germany) without sodium bicarbonate, with phenol red, 2 mM stable glutamine, and supplemented with 10% heat-inactivated foetal bovine serum (Biochrom, Berlin, Germany), 100 U/ml penicillin, 100 μg/ml streptomycin, 10 μg/ml gentamicin, and 2.5 μg/ml amphotericin B (all from Sigma-Aldrich, St. Louis, MO) were used. The second medium tested, designated medium B, was RPMI 1640 medium (Lonza, Verviers, Belgium) containing 25 mM sodium bicarbonate, 2 mM stable glutamine and phenol red was supplemented with 50 μM 2-ME (Gibco, New York, NY), 10% heat-inactivated foetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 μg/ml gentamicin, and 2.5 μg/ml amphotericin B.
The other chemicals used for the assays were 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, AppliChem, Darmstadt, Germany), Trypan blue solution (Biochrom, Berlin, Germany), Dimethyl sulfoxide (DMSO, Invitrogen/Thermo Fisher Scientific, Carlsbad, CA), 2′,7′-dichlorodihydrofluorescein diacetate) (H2DCFDA, Invitrogen, Carlsbad, CA), Neutral Red, 2,2′-azo-bis-(2-methylpropionamidine) dihydrochloride (AAPH) 1 M stock solution, 123-Rhodamine. The Griess reagent was prepared from 5% orthophosphoric acid, 1% sulphanilamide, and 0.1% N-(1-Naphthyl) ethylenediamine dihydrochloride standard was NaNO2 (the following chemicals were purchased from Sigma-Aldrich, St. Louis, MO).
Other chemicals including NH4Cl, EDTA, and sodium azide were used for cell biology applications. Methanol with purity of 99.99% (HiPerSolv, Chromanorm, Fontenay-sous-Bois, France) from VWR was used for HPLC analysis of astaxanthins. H. pluvialis biomass was purchased from Algamo, s. r. o. (Mostek, Czech Republic).
Preparation of oleoresin from Haematococcus pluvialisFor the preparation of oleoresin from H. pluvialis, 60 g of dry biomass of the microalga was used, which was extracted with 600 ml of acetone. The extraction process was assisted by sonication with an ultrasonic bath (K6 Kraintek, s.r.o., Podhájska, Slovakia) with a frequency of 38 kHz and an intensity of 47.7707 W/cm at 25°C and for 30 min. The resulting suspension was centrifuged to facilitate the removal of suspended particles. The extraction procedure was repeated three times with the residual biomass. The supernatant obtained from the centrifugation of the extraction procedures was combined and evaporated under reduced pressure using a rotary evaporator at a temperature of 30°C. 22.05 g of oleoresin was obtained, which was used for biological evaluation.
HPLC-APCI-HRMS analysis of the oleoresin from H. pluvialisAnalysis of H. pluvialis oleoresin was performed using a Dionex UltiMate 3000 HPLC system (Thermo Scientific, Sunnyvale, CA) coupled to a high-resolution tandem mass spectrometry (HRMS/MS) detector with atmospheric pressure chemical ionization (APCI) source (Impact HD mass spectrometer Bruker, Billerica, MA) (HPLC-APCI-HRMS). A Luna® C8 column (100 × 4.6 mm, 3 μm, Phenomenex) maintained at a temperature of 30°C was used to separate the target compounds. The mobile phase consisted of water (A) and methanol (B), both with 0.1% formic acid to increase ionization efficiency. An elution gradient of the mobile phase with a flow rate of 0.8 ml/min was applied as follows: 0–20 min, 20–0% A; 20–25 min, 0% A; 25–27 min, 0–20% A; 27–30 min, 20–20% A. The conditions for MS analysis were as follows: dry temperature 250°C; drying gas flow 12 l/min; nebulizer 3 bar; the voltage of the spray needle 4.2 kV. Mass spectra were recorded in positive ionization mode in the range of 50–2000 m/z at a scanning rate of 2 Hz. Collision energy of 35 eV was used, with nitrogen serving as the collision gas. Sodium formate clusters were used for spectral calibration at the beginning of each analysis. The Smart Formula function in Bruker Compass DataAnalysis software (version 4.2) was used to calculate the formulas for the molecular peaks and fragments. Identification of individual oleoresin peaks was determined by comparing data from HPLC-APCI-HRMS with existing literature data. The HPLC analysis of oleoresin is shown in Supplementary Fig. 1.
Isolation and cultivation of mouse spleen cellsMale and female Balb/c mice were originally purchased from Velaz (Prague, Czech Republic) and bred in at the animal facilities of Institute of Parasitology of the Slovak Academy of Sciences under pathogen-free conditions. Spleens were aseptically isolated from 2 to 3 male mice in each experiment. Cell suspensions were obtained by passing spleen tissue through 40 μm nylon filters (BD Biosciences, Darmstadt, Germany) in 5 ml of cold culture medium A. Red blood cells were removed by incubating the suspension with 5 ml of lysis solution (8.02% NH4Cl, 0.85% NaHCO3, and 0.37% EDTA) for 5 min on ice. After centrifugation, splenocytes were washed twice in PBS and filtered through nylon filters (BD Biosciences). Mouse splenocyte suspensions were pooled, counted, and analysed for viability using the trypan blue exclusion assay. Then, the cells were resuspended in medium A or medium B for the experiments. The study was performed in accordance with the Guidelines for the Care and Use of Experimental Animals, No. 289/2003, in the Slovak Republic. The experiment was approved by the Ethics Committee of the State Veterinary and Food Administration of the Slovak Republic.
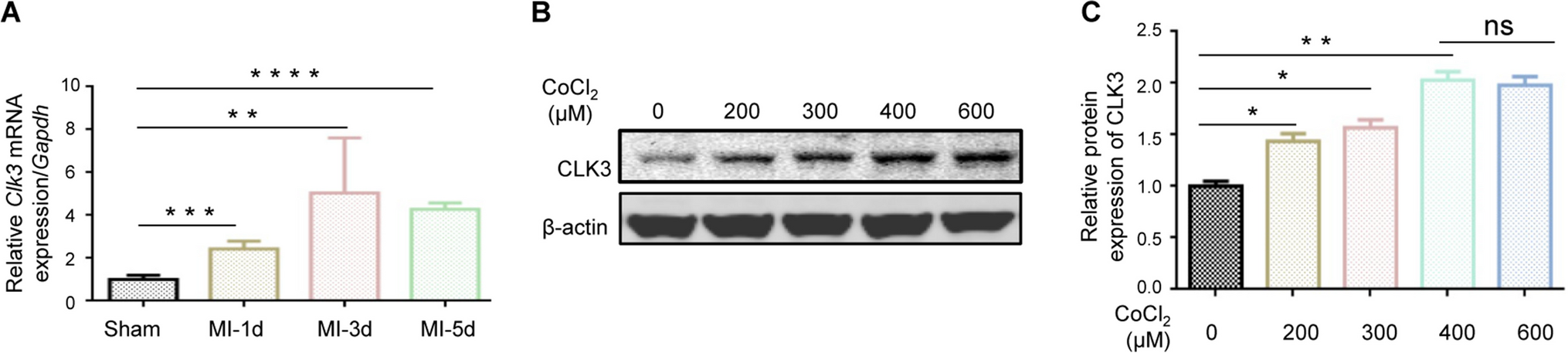
Experimental designIn order to obtain a first insight whether standard culture conditions can affect splenocyte viability after 24 h of incubation, an experiment was designed using cells cultured in two media that differ in free radical reduction conditions (Fig. 1). In addition, oxidative stress was induced in the cells by a water-soluble molecule 2,2′-azo-bis-(2-methylpropionamidine) dihydrochloride (1 mM) for 1 h in the assays to monitor the level of ROS. The AAPH system stimulates the production of free radicals at physiological temperature without generating H2O2 as an intermediate, thereby achieving an independent effect from the enzymatic antioxidant system of cells. Within these experimental settings, we assessed viability, metabolic activity, neutral red absorption, mitochondrial membrane potential, expression of genes controlling endogenous antioxidant mechanisms, intracellular ROS production, and nitrite oxide production. To determine how the influence of culture conditions is modulated by the addition of oleoresin (OLR) with significant antioxidant effect, we also repeated all tests after addition of OLR to splenocytes in both types of culture media.
Figure 1.
Created with BioRender.com
Trypan blue exclusion (viability test)Splenocyte suspension diluted to a concentration of 1 × 106 cells/ml in medium (A or B) was placed on CultureSlides (Falcon Tissue Culture Treated Glass Slides, Corning, Glendale, AZ) in triplicate for each treatment. OLR working solution was added to the cells in a concentration-dependent manner. Splenocytes were incubated for 24 h at 37°C, 5% CO2, humidity, and atmospheric oxygen in medium A and B, respectively. Untreated cells in medium A and B were used as controls, and naïve, freshly isolated cells were used as intact controls. Trypan blue (10% solution, 10 µl) was added to the wells, and the proportions (%) of live cells to total cells/well counted (for a total of 300 cells) were calculated for the control sample and each OLR concentration. Final data are given as mean ± SD. Viability was assessed and verified in two independent in vitro experiments.
MTT assayThe MTT assay is a colorimetric test for the evaluation of cellular metabolic activity based on the conversion of the water-soluble yellow dye MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) to insoluble purple formazan by the action of mitochondrial and cytoplasmic reductases (Berridge and Tan 1993; Kumar et al. 2018). A suspension of splenocytes diluted to a concentration of 1 × 106 cells/ml in medium (A or B) was placed in 24-well plates (Corning, Glendale, AZ) in triplicate for each treatment and control, and cells were then subsequently incubated for 24 h under the same conditions as described above. Untreated cells in both medium A and medium B were used as control and naïve, freshly isolated cells as intact control. OLR working solution was added to the cells in a concentration-dependent manner. MTT (5 mg/ml in PBS) was added to the cell suspension 4 h before end of the assay in a volume of 50 μl/ml. The non-adherent cells were transferred to tubes, and the adherent cell population was washed with PBS. Then, 100 µl of DMSO was added to each well and to the tubes containing the non-adherent cells. OD values for adherent and non-adherent cells/sample were summed and used to calculate the mean ± SD. The in vitro experiment was repeated three times for spleen cells cultured in both medium A and B.
Neutral red uptake assayThe principle is the ability of living cells not only to incorporate, but also to bind the dye neutral red in response to the pH gradient through the production of ATP in lysosomes, and this after only a short period of co-cultivation (Repetto et al. 2008). A suspension of splenocytes diluted to a concentration of 1 × 106 cells/ml in medium (A or B) was placed in 24-well plates in triplicate for each treatment and control. Untreated cells in medium A/B were used as control and naïve, freshly isolated cells as intact control. OLR at selected concentrations was added to the cells, and splenocytes were incubated for 24 h under standard conditions. One hour before the end of cultivation, 20 µg/10 µl of neutral red was added to each well. Dye extraction was performed separately for non-adherent and adherent cells using 200 μl of extraction buffer (1% glacial acetic acid, 50% ethanol in distilled water). Optical density of extracts from both cell counterparts/wells was measured in a 96-well plate at 550 nm using a Multiscan FC Plate Reader. Two in vitro experiments were performed.
Mitochondrial membrane potentialMitochondrial membrane potential reflects the process of electron transport and oxidative phosphorylation, which is why it is considered a key indicator of mitochondrial activity (Chao et al. 2017). Loss of mitochondrial potential indicates bioenergetic stress and subsequently leads to the release of apoptotic factors that result in cell death (Gutierrez et al. 2017). Splenocytes diluted to a concentration of 1 × 106 cells/ml in medium A or B were treated with selected concentrations of OLR in 24-well plates for 24 h. Naive cells were used as intact control. Untreated cells in medium A or B were used as control. Mitochondrial membrane potential (Δψm) was determined by flow cytometry using the fluorescent dye rhodamine 123, which is absorbed by mitochondria of living cells. Changes in dye absorption reflect Δψm and are expressed as mean fluorescence intensity (MFI). After vigorous resuspension, splenocytes were transferred to test tubes and a dye was added at a final concentration of 10 µM. Cells were incubated at 37°C for 20 min, then centrifuged, the supernatant was removed, and cells resuspended in PBS were immediately used to measure Δψm. Flow cytometry was performed using a FACS Canto flow cytometer with rhodamine 123 excitation at 505 nm and emission at 535 nm. The experiment was performed three times.
Annexin V/propidium iodide apoptosis assayThe apoptosis assay allows detection of changes in cells associated with programmed death and then quantification of cells undergoing apoptosis, which allowed us to evaluate the effects of the culture environment as well as the influence of the extract tested. Splenocytes were diluted to a concentration of 1 × 106 cells/ml in medium A or medium B, plated in plates with 24 wells, and treated with OLR (10 and 40 µg/ml) for 24 h. Untreated cells were used as control and naïve cells as intact control. After the incubation period, the non-adherent splenocyte population was transferred to tubes, and the adherent cell population was detached with 300 µl of warm Accutase Cell Detachment Solution (BioLegend, San Diego, CA) for 20 min at 37°C, collected into tubes, and washed. Then, both cell fractions/wells were pooled, washed in cold PBS, and stained at room temperature with Annexin V and propidium iodide solutions using the BD Pharmingen Annexin V-FITC apoptosis detection kit (BD Biosciences, San Jose, CA) according to the manufacturer’s instructions. Analysis was performed by flow cytometry using a FACS Canto flow cytometer. The percentages (%) of live cells and cells in different stages of apoptosis were evaluated using FACS Diva software. The assay was performed twice.
Determination of the amount of nitric oxideSuspensions of splenocytes diluted to a concentration of 1 × 106 cells/ml in medium A or B were plated in 24-well plates in triplicate/treatment types and treated with OLR (10 and 40 µg/ml) for 24 h. Untreated cells in medium A/B were used as control, and freshly isolated (naïve) cells were used as intact control group. Two hours before the end of cultivation, splenocytes were treated with AAPH (10 mM) to induce nitric oxide (NO) production. The concentration of NO in the supernatant was measured as nitrite (NO2−) using the Griess reagent in a 96-well plate (in triplicate). Absorbance was measured at 550 nm using a Multiscan FC Plate Reader. Nitrite concentration was determined from a calibration curve using 0.1 M NaNO3 as a standard.
Determination of intracellular reactive oxygen species in splenocytesSplenic cells diluted in medium A or medium B to a concentration of 1 × 106/ml were plated in 24-well plates in triplicate/treatment and treated with OLR (10 and 40 µg/ml) for 24 h. Untreated cells in both medium A and B were used as control and naïve, freshly isolated cells as intact control. The production of intracellular reactive oxygen species was determined using the fluorescent dye H2DCFDA. The dye was added at a concentration of 1 mM 4 h before the end of incubation. To induce oxidative stress in cells, AAPH (10 mM) was added to cell cultures 1 h before the end of the assays. Non-adherent cells were transferred to tubes; adherent splenocytes were detached with 300 µl of warm Accutase for 20 min at 37°C and then transferred to tubes and washed. Both cell fractions from each individual well were pooled and resuspended in 200 µl of culture medium (A or B). The percentage of cells producing ROS and the mean fluorescence intensity (MFI) corresponding to the concentration of ROS were measured by flow cytometry using a FACS Canto flow cytometer. The assay was repeated twice for both culture media.
RNA isolation from splenic cells and quantitative RT-PCR analysisCell suspensions (1 × 106/ml) diluted in medium A or B were placed in 24-well plates (Corning, NY) in triplicate for each treatment and treated with 10 or 40 µg/ml OLR concentration for 24 h. Untreated cells in medium A and B were used as control. Naïve, freshly isolated cells were used as intact control. Supernatants containing lymphocytes were collected in 1.5-ml tubes, and cell pellets were immersed in 0.5 ml of Trizol reagent (Invitrogen). Adherent cells were immersed in 0.5 ml of Trizol, and both cell fractions/wells were pooled and used for RNA extraction. RNA was quantified using an AstraGene Nanospectrophotometer (Harston, Cambridge, UK), and then, 2 µg was transcribed into cDNA using ReverseAid H minus M-MuLV reverse transcriptase and oligodT primers (Thermo Fisher Scientific, Burlington, Canada). The cDNA for each sample was used as a template for quantitative PCR. RT-PCR analysis of relative mRNA abundance was determined using SYBR green master mix (Sigma-Aldrich, St. Louis, MO) on a BioRad CFX thermocycler (BioRad, Hercules, CA). RT-PCR was performed in 20 μl reactions. The list of primers from the study by Wang et al. (2019) used in qRT-PCR is summarized in Table 1. The in vitro experiment was repeated twice.
Table 1. Primers used for gene expression analysis by qRT-PCR Statistical analysisAll data were calculated as means ± standard deviation (SD) from two or three independent in vitro experiments. Statistical analysis of data was performed using GraphPad Prism (version 7) for Windows (GraphPad Software, Inc., San Diego, CA). Results were analysed using either one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test or grouped analysis using two-way ANOVA and Sidak post hoc test. Statistically significant differences were calculated for *p < 0.05, **p < 0.01, and ***p < 0.001.
留言 (0)