
Remember me
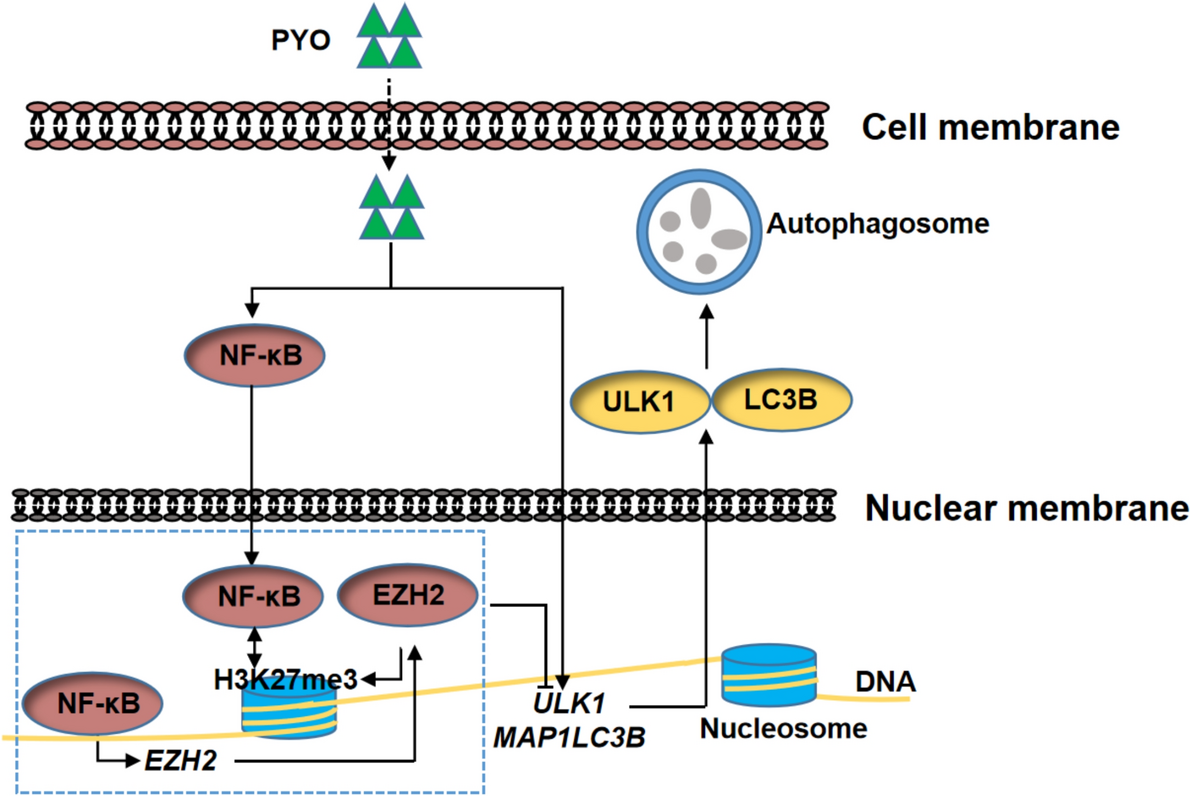
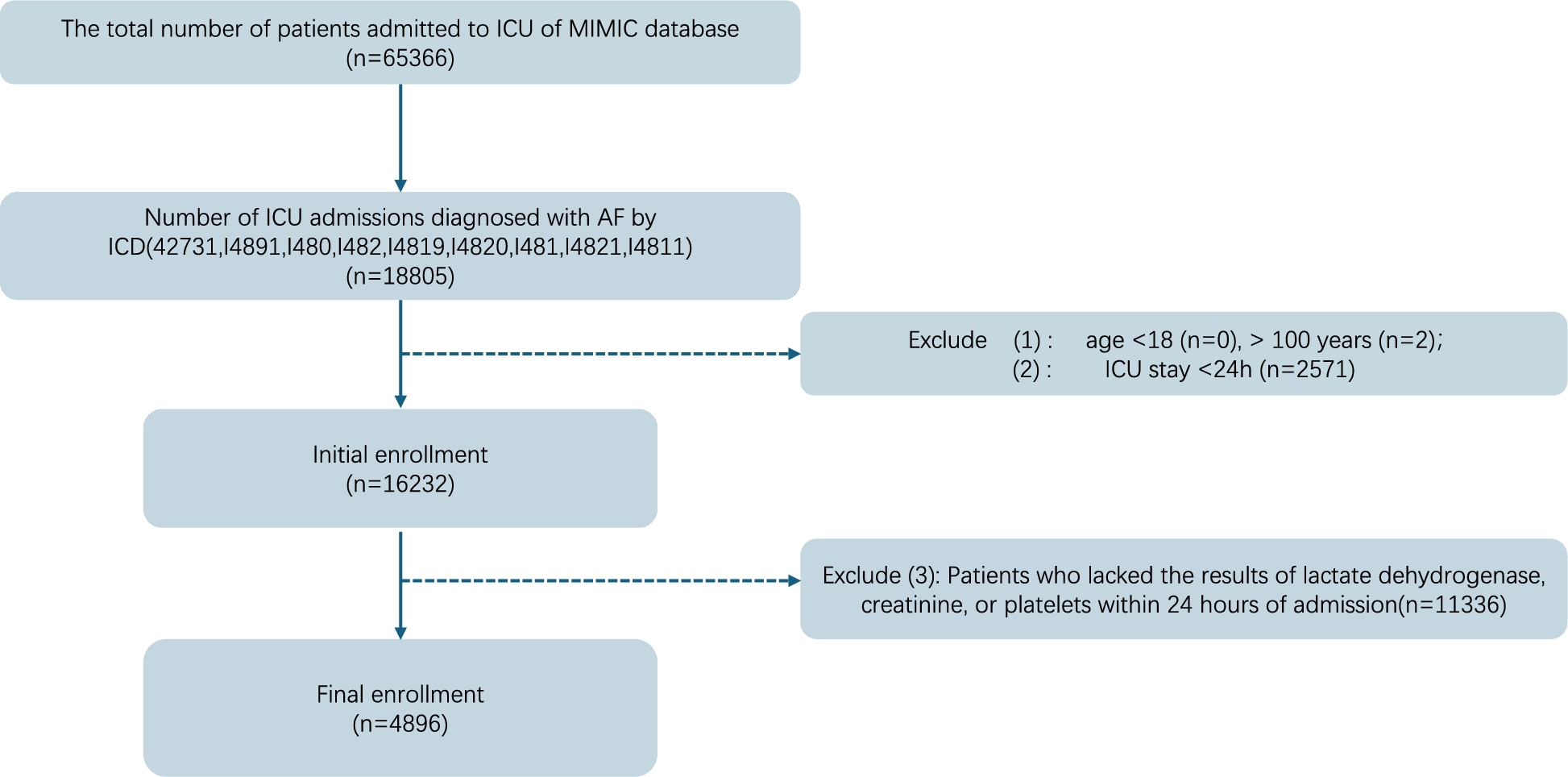






To understand the DRGs set associated with HCC, “limma” method was utilized to identify DEGs between normal tissues and tumor tissues. | log2FC |> 1.5 was considered as significantly differentially expressed genes. Finally, we get 229 downregulated genes and 161 upregulated genes. Volcano maps and heat maps were generated to show the final results (Fig. 2A, B). In addition, our previously collected 2774 DRGs were intersected with the above 390 DEGs, resulting in 45 co-expressed genes. Venn diagram was used to visualize the 45 co-expressed genes (Fig. 2C). To further understand the interconnections between the 45 genes, the spherical network diagram was generated to display the correlation between each gene (Fig. 2D).
Fig. 2
Identification of co-expressed genes. A Volcanic plot of DEGs in tumor and normal tissues of HCC patients. B Heatmap of 30 top representative DEGs in HCC patients. C A Venn diagram is generated to represent 45 co-expressed genes of DRGs and DEGs. D Protein interaction network diagram of 45 co-expressed genes
Consensus clustering associated with 45 co-expressed genesAim to explore the relationship between co-expressed genes and HCC subtypes, we applied k-mean algorithm to divide HCC patients into two separate clusters (C1 and C2) (Fig. 3A). The cumulative distribution function (CDF) and the relative change under the CDF curve suggested that k = 2 was the best clustering result (Fig. 3B, C). Additionally, heat maps that display the expression of 45 co-expressed genes in the two subgroups were created. Results indicate that some genes, such as ANGPTL6, AGXT2, RDH16, C8A, GYS2, ACSM5, FBP1, ALDH8A1, BHMT and SPP2 are highly expressed in C2. The expression of some genes, such as SFN, CDKN2C, CENPW, RRM2, CDCA8, E2F1, RNASEH2A, CDT1, CENPM, ADAM15, and others, is noticeably higher in C1 cluster (Fig. 3D). To further verify the mutual independence between the two clusters, PCA was powered to verify the two subgroups. The results suggested significant separation between the two subgroups (Fig. 3E). Moreover, C1 subgroup indicated poor prognosis according to the prognosis curve of overall survival in Fig. 3F.
Fig. 3
Consensus clustering based on 45 co-expressed genes. A Clustering heatmap of co-expressed genes. B Cumulative distribution function area of different clustering (k = 2–10). C The relative change of area under the CDF curve. D An heatmap of 45 co-expressed genes in different clustering groups. E PCA plot of two clustering subgroups. F Analysis of overall survival between different cluster subgroups
Enrichment of biological functions and signaling pathwaysTo confirm whether consensus clustering result related to disulfidptosis, “limma” method was measured to identified DEGs between two subgroups, then the DEGs with | log2FC |> 1 were used for GO and KEGG enrichment analysis. Bubble maps were generated to visualize signaling pathways, biological processes, cell composition, and molecular functions, respectively (Fig. 4A–D). From the results, different cluster subgroups were observed to be involved in complement and coagulation system, metabolism of cytochrome P450, drug metabolism, bile secretion and retinol metabolism, PPAR signaling pathway, chemical carcinogens–DNA adducts. Meanwhile, the cellular response to xenobiotic stimulation, iron ion binding, biological oxidative stress and lipoprotein particles, endoplasmic reticulum lumen, and granular lumen were also involved in two different subgroups. Complete GO and KEGG analysis are summarized in Additional file 3. To further investigate the relevant signaling pathways of DEGs, we performed GSEA enrichment analysis of them. The results showed that the DEGs between two cluster subgroups were mainly involved in cell cycle and metaphase, oxidative stress and retinol metabolism. Mountain map, bubble plot, GSEA classic plot and bar chart were generated, respectively, for comprehensive visualization of GSEA enrichment analysis results (Fig. 4E–H). The full GSEA enrichment results are presented in Additional file 4.
Fig. 4
Signaling pathways and functional enrichment analysis between different subgroups. A–D Representative bubble plot analyzed by KEGG and GO. Bubble plots representing KEGG (A), BP(B), MF (C) and CC (D) are generated, respectively. E, H Visualization of representative results analyzed by GSEA. Visualization of mountain plot (E), bubble plot (F), classic plot (G) and bar plot (H) for GSEA analysis results
Establishment of prognostic risk factor modelIn order to further screen genes related to prognosis, we used LASSO Cox regression analysis for extraction. Seven candidate genes that met the minimum lambda value of 0.045532 were screened out. Partial likelihood deviance plots and coefficients distribution curves were generated to visualize the LASSO Cox regression results (Fig. 5A, B). A volcano plot was utilized to show the location of the seven candidate genes in the differential gene ranking (Fig. 5C). In order to further ensure the reliability of core genes, univariate and multivariate Cox regression analysis were used to further screen three hub genes (RDH16, SPP2 and CDCA8). The forest plot is shown in Fig. 5E, F. Heatmap of three hub genes related to prognosis is shown in Fig. 5D.
Fig. 5
Establishment of prognostic risk factor model. A, B) Partial likelihood deviation plot and coefficient curve of LASSO Cox analysis. C A DEGs sequenced heatmap of 7 genes extracted by LASSO Cox regression analysis. D A heatmap of prognosis-related risk factors and risk score model. E–F Forest plots of univariate (E) and multivariate Cox regression (F)
Risk score correlates with somatic mutations and overall survivalTo investigate the relationship between risk score and prognosis, we divided the cohort into high and low risk score subgroups based on the median risk score. KM curves were generated to compare the differences in overall survival between the two risk score subgroups. The results suggested that high risk score predicted adverse prognosis (Fig. 6A). Additionally, we found similar results in the LIRI-JP&GSE76427 verification set and there was statistically significant difference (P < 0.001) (Fig. 6B). To further confirm the accuracy of the risk assessment model, we used ROC curve and line graph to evaluate the reliability of the risk prognosis model. The results indicate that the AUC values of 1-, 3-, and 5-year predictions of this model are, respectively, 0.752, 0.695, and 0.691, indicating that this model has a certain reliability (Fig. 6C, D). In order to observe the somatic mutation difference between the high risk score subgroup and the low risk score group, mutation data of the TCGA-LIHC training set were collected and a waterfall diagram was generated (Fig. 6E). The results showed that missense mutation accounts for the majority. In addition, TP53 (32.4%), TTN (28.6%) and CTNNB1 (27.6%) were the genes with the top three largest number of mutations in all somatic mutation.
Fig. 6
Somatic mutations and prognosis in different risk score subgroups. A, B KM curves of overall survival of different risk score subgroups in TCGA training set and LIRI-JP & GSE76427 verification set. C, D ROC curve (C) and broken line plot (D) of risk score related 1-, 3-, and 5-year overall survival prediction. E Waterfall plots of somatic mutations in different risk score subgroups
Risk score is correlated with clinical characteristicsTo visualize the risk score of HCC patients with different pathological characteristics, violin plots were generated. The results showed that there was a positive correlation between tumor diameter and risk score in HCC patients (Fig. 7A). Similarly, we found that tumor stage and tumor grade were also positively correlated with risk score (Fig. 7B, C). However, there was no significant difference in risk score between patients with lymph node or distant metastasis in HCC, which may be due to the small sample size (Fig. 7D, E). In addition, we also generated the clinical prognosis of patients with different pathological features in HCC. The results suggested that the prognosis of HCC in the high risk score subgroup was worse than that in the low-subgroup group in different stages, grade and tumor volume (Fig. 8A–F). Similarly, high risk score indicated unfavorable prognosis when there was no lymph node metastasis or distant metastasis (Fig. 8G, H).
Fig. 7
Risk score is related to the clinical characteristics of HCC patients. A–E Violins of risks core and tumor size (A), clinical stage (B), clinical grade (C), distant metastasis (D), and lymph node metastasis (E)
Fig. 8
Risk score is associated with the prognosis of HCC patients with different clinical characteristics. A, B KM curves of different tumor size in different risk score subgroups. C, D KM curves of different clinical grades in different risk score subgroups. E, F KM curves of different clinical stages in different risk score subgroups. G KM curves of HCC patients without lymph node metastasis in different risk score subgroups. H KM curves of HCC patients without distant metastasis in different risk score subgroups
Tumor immune analysis of different risk score subgroupsTo observe the infiltration levels of tumor-associated immune cells in different risk score subgroups, we used the CMP counts algorithm to calculate 10 tumor-associated immune cells. Stacked plot and bar graph were generated for visualization of the various immune cells (Fig. 9A, B). To verify the above results, we used CIBERSORT algorithm to re-evaluate the differences between different subgroups of immune cells (Additional file 1: Fig. S1). The results showed that regulatory T cells (Tregs) and CD4 + T cells were significantly enriched in the high-risk subgroup. Moreover, the ESTIMATE algorithm was applied to calculate somatic score, immune score and ESTIMATE score of two subgroups. The results showed that the stromal score of high risk score group was lower than that of low risk score group, but there was no significant difference between immunological score and ESTIMATE score (Fig. 9C). Aim to further investigate the association of immune cells with risk score, we utilized scatter plots for visualization. The results suggested that neutrophils, CD4 T cells, dendritic cells, and B cells were positively correlated with the risk score (Fig. 9D–G).
Fig. 9
Tumor immune analysis of different risk score subgroups. A A stack diagram of 10 types of immune cells. B CMP counter score of 10 tumor-associated immune cells in different subgroups. C Violin plots of somatic score, immune score and ESTIMATE score between risk score subgroups. D–G Scatter plots of risk score correlation with neutrophils, CD4T cells, dendritic cells, and B cells. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
Prediction of response to immunotherapy and chemotherapeutic drug reactivityTo understand the responsiveness of HCC patients with different risk score subgroups to immunotherapy, we also generated violin plots to compare 8 immune checkpoint inhibitor (ICIs) genes and 20 HLA-related genes [14]. The results suggest that, most of the high risk score group had ICIs-related genes (CDC274, CTLA4, HAVCR2, TIGIT and PDCD1) and HLA-associated genes (CEP112, CEP68, AASS, CENPF, NUP210, ANO6, CLIP4, FBLN5, ATP6AP1, PRKAR2B, CHPF, MYCBP2, NAT14, SLC9A3R1, TMEM97 and MFGE8) were upregulated, indicating that the high risk score group may have a poor response to immunotherapy (Fig. 10A, B).
Fig. 10
Prediction of response to immunotherapy. A, B Bar charts of ICIs-related genes (A) and HLA-related genes B in different subgroups. C Violin charts of risk score in HCC patients who did and did not respond to immunotherapy. D–F Correlation scatter plot of risk score with TIDE score (D), exclusion score E and dysfunction score (F). (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001)
To further explore whether the response of HCC patients to immunotherapy can be predicted by the risk score, we used the TIDE database to predict the samples of tumor patients in the TCGA-LIHC cohort. The results dedicated that the response to immunotherapy in the high risk score group is worse than low risk score group (Fig. 10C). In addition, the result show that risk score is positively correlated with TIDE score as well as exclusion score (Fig. 10D, E), while negatively correlated with dysfunction score (Fig. 10F). "pRRophetic" package was powered to predict the half maximal inhibitory concentration (IC50) of HCC-related anti-cancer drugs. The result showed that HCC patients in the high risk score subgroup were more sensitive to gemcitabine, vinorelbine and paclitaxel (Fig. 11A–C). The ROC curve showed that the predictive value of IC50 of gemcitabine had a certain accuracy (AUC = 0.765), while the predictive value of IC50 of vinorelbine and paclitaxel was uncertain (AUC < 0.7) (Fig. 11D–F). The above results suggest that gemcitabine may be an effective therapeutic agent for the treatment of HCC in the high risk score subgroup.
Fig. 11
Prediction of response to chemotherapeutic drug reactivity. A-C IC50 bar plots of gemcitabine (A), vinorelbine (B) and paclitaxel (C) among different high-risk and low-risk subgroups. D–F IC50-related ROC curves of gemcitabine (D), vinorelbine (E), and paclitaxel (F). (****p < 0.0001)
Construction of prognostic models related to risk factorsIn order to further elaborate the relationship between independent risk factors and the prognosis of HCC patients, a nomogram was generated to construct a prognostic prediction model (Fig. 12A). Based on the clinical characteristics and hub genes of each individual HCC patient, the overall risk score was calculated, and then the probability of survival after 1-, 3-, and 5 years was calculated. In addition, calibration plots were established to test the reliability of the prediction model (Fig. 12B). The results showed that the 1-, 3- and 5-year survival rates predicted by the model were close to the ideal line, indicating that the prediction model was reliable. Similarly, DCA curves were generated to see the effectiveness of each independent risk factor (Fig. 12C). Since each independent factor does not intersect the two slash lines, it indicates that the above independent factors are valid. To further determine the association of different independent risk factors with prognosis, univariate Cox regression and multivariate Cox regression analyses were used to visualize clinical characteristics. The results showed that different pathological features (TNM) and risk score were significantly associated with prognosis (Fig. 12D, E).
Fig. 12
Construction of prognostic model related to risk factors. A A graph of independent risk factors associated with prognosis. B, C Calibration plot and DCA plot for risk score model correlation. D, E Univariate and multivariate Cox regression forest plots for prognosis-related risk factors
Validation of hub genes at the methylation and total protein levelsTo further investigate the expression of hub genes in HCC, we downloaded the immunohistochemical results of the corresponding hub genes from the Human Protein Atlas (HPA) database. In addition, the total protein expression levels of hub genes were collected in UALCAN database to validate the results. The results showed that CDCA8 was highly expressed and RDH16 was lowly expressed in HCC tumor tissues (Additional file 1: Fig. S2). Unfortunately, we did not find SPP2 results in the HPA database. Furthermore, we downloaded the methylation data of CDCA8, SPP2 and RDH16 in the UALCAN database. The results showed that CDCA8 (Additional file 1: Fig. S3) and SPP2 methylation levels were decreased in tumor tissues with worse clinical stages (Additional file 1: Fig. S4). There was no significant difference in the methylation level of RDH16 (Additional file 1: Fig. S5).
Comments (0)