
記住我


Classical Hodgkin lymphoma (cHL) is a B cell neoplasm, accounting for 10% of all lymphomas and <1% of all cancers.1 The incidence rate is 2–3 people per 100,000 population per year with a peak incidence in 20–34 years.2 Despite advances in the treatment of cHL over the past 20–30 years, when using standard chemotherapy protocols (ABVD/esBEACOPP) in combination with radiotherapy, still 10%–20% of patients with early stages and 35%–45% with late stages of the disease relapse or are refractory to first-line therapy, respectively.3 Younger patients with refractory/relapsed cHL (r/r cHL) are eligible for autologous hematopoietic stem cell transplantation (auto-HSCT), 50% of whom relapse or are refractory to this type of treatment.4 The introduction of new drugs, such as the Brentuximab vedotin5 and anti-PD1 antibodies (Pembrolizumab, Nivolumab)6 have significantly improved treatment outcomes in patients with r/r cHL as well as in newly diagnosed cHL patients.
New approaches in the treatment of cHL include the use of Bruton’s tyrosine kinase inhibitors, JAK2 inhibitors, immunomodulatory drugs (lenalidomide, etc.), an immunoconjugate of a monoclonal antibody to CD25 with cytotoxin pyrrolobenzodiazepine (Camidanlumab tesirine), a bispecific antibody to CD16 and CD30 (AFM13) and adoptive cellular immunotherapy (including Chimeric antigen receptor T cell [CAR-T cell] therapy).7
Following advances in clinical trials and FDA approval of anti-CD19 CAR-T cell therapies for non-Hodgkin’s lymphomas and B-ALL, as well as an anti-B-cell maturation antigen (BCMA) CAR-T cell therapy for multiple myeloma, there is a growing interest in the scientific community to explore the potential of CAR-T cell therapy in cHL. CAR-T cell technology overview is out of the scope of this article, it is discussed elsewhere.8
cHL PATHOGENESIS AND NEOPLASTIC PATHOLOGYTransformed mature B cells are the tumor substrate of cHL, constituting <1% of the entire tumor mass. Monoclonal large bi- or multinuclear Reed-Sternberg cells, Hodgkin mononuclear cells (Hodgkin and Reed-Sternberg [HRS]), and mummified cells (condensed cytoplasm and pyknotic eosinophilic or basophilic nuclei) are pathognomonic for the disease.9 These cells express CD30 in >98% of cases, CD15 in 75% of cases, CD20 in 30%–40% of cases, PDL1 in 80% of cases, PDL2 in 40% of cases10 and also in most cases are weakly stained for PAX5 antigen.11,12 The expression of Bcl2, p53, and PCNA by HRS cells correlates with chemoresistance.13,14
On histological examination of biopsy material, tumor cells do not express CD45, EMA, or other markers of B cell lineage differentiation (Oct-2, BoB.1, and B cell receptor).15,16
The primary oncogenic event occurs in B cells of the germinal center and all HRS cells have the same monoclonal rearrangement of heavy chain variable domain genes that have passed the stage of somatic hypermutation.17 Under normal physiological conditions, B cells with defective expression of the high-affinity B cell receptor undergo apoptosis. However, in the context of oncogenic transformation, HRS cells evade cell death by sustaining the activation of the NF-kB pathway through acquired mutations in negative regulators of the pathway, such as IκB and A20 proteins.18–20 Additionally, alterations in components within the NF-kB pathway itself could contribute to the sustained activation of NF-kB signaling.21 In cases of Epstein-Barr virus (EBV)-associated cHL, the expression of LMP1/2A proteins further supports cell survival.22 Furthermore, disruption of epigenetic regulation in HRS cells leads to the loss of key transcription factors responsible for B cell differentiation.11
The vast majority of patients with cHL are characterized by chromosomal abnormalities of region 9p24.1. This region encompasses the PDJ amplicon genes, including PDL1 (CD274), PD-L2 (PDCD1LG2) and JAK2.23 In the study of Romer et al, the following changes in the 9p24.1 locus were detected in primary cHL patients: polysomy (5%), copy number gain (58%) and amplification (36%) of the PD-L1 and PD-L2 loci.24 Such changes lead to increased expression of PDL1, PDL2, and JAK2.25 More than 87% of patients demonstrate deregulation of the JAK-STAT pathway. Activating mutations in the JAK1, STAT3, STAT5B, and STAT6 genes, which are frequently accompanied by inactivating mutations in the SOCS1 gene, can directly contribute to this.26
The tumor microenvironment (TME) of cHL is represented by reactive T and B lymphocytes, histiocytes, neutrophils, eosinophils, basophils, plasma cells, dendritic cells, as well as fibroblasts, reticular cells, and endothelial cells.27
HRS cells manipulate the cellular composition of TME by producing various chemokines to attract lymphocytes (mainly Tregs) (CCL17/TARC, CCL-5, CCL-20, and CCL22), macrophages, neutrophils, eosinophils and basophils (IL-5, IL- 8, IL-9, CCL-5, and CCL-28), which in turn produce chemokines such as CCL-3, CCL-4 and CCL-8.28,29 HRS cells ligation of CD40 and CD30 with the corresponding ligands on TME cells, leads to the transmission of proliferative and anti-apoptotic signals to the tumor cells.30,31 HRS cells produce a range of cytokines such as IL-6, IL-7, IL-8, IL-13, and TNFa, PGE2, and TGFβ, which support the expansion of TME cells, in addition, providing anti-apoptotic and proliferative signals in autocrine and paracrine manner. CD68+ macrophages and Treg cells in turn produce a number of cytokines such as IL-10, TGFb, and others, resulting in the polarization of TME into an immunosuppressive state and inhibition of the immune response.32 Another important feature of immune evasion in cHL cells is the genetically determined overexpression of PD1 ligands. In clinical studies, monoclonal anti-PD1 antibodies (nivolumab, pembrolizumab) have been able to achieve an overall response and complete remission in patients with r/r cHL in approximately 70% and 30% of patients, respectively.25
In >90% of patients with cHL, HRS cells have reduced expression of MHCI33 and approximately 40% show decreased expression of MHCII, which leads to defective immune surveillance by CD8 and CD4 T cells, respectively.34 MHCI reduced expression is in most cases due to mutations in the B2M gene.33 This finding has prompted researchers to consider CAR-T cell therapy as a promising and immunologically justified therapeutic modality for the treatment of r/r cHL.
KEY TARGETS FOR CAR-T THERAPY IN cHLCD30 (TNFRSF8) is a type 1 transmembrane glycoprotein of the superfamily tumor necrosis inflammatory receptors with a molecular weight ranging from 105 to 120 kDa.35 CD30 is a major marker of HRS cells and its expression occurs in >95% of cases of cHL.36,37 Signal transduction through CD30 leads to the activation of NF-kB and mitogen-activated protein kinase (MAPK) pathways, resulting in proliferative and anti-apoptotic effects in HRS cells.38 At the same time, under physiological conditions, CD30 expression is found on minor subpopulations of activated B lymphocytes, and NK cells, as well as on 3%–31% of circulating T lymphocytes.39 During viral infection, the number of CD30+ cells in the peripheral blood can reach 95% on the third day of infection.40 IL-4 and CD28 signaling are required for CD30 expression on T cell surfaces.41 A low level of CD30 expression can be observed in the cells of the adrenal gland and pancreas.42,43 Transcription factors Sp1 and JunB are responsible for the induction of CD30 expression in cHL.44,45 Promoters of CD30 gene contain CpG islands with low-methylation features that are related to the increased CD30 expression in cHL.46 A soluble extracellular 88 kDa CD30 protein can be detected in the blood of patients with cHL. It is generated through the cleavage of the supramembrane region of the receptor by metalloproteinases, such as ADAM10 and ADAM17.38,47
Brentuximab vedotin, a CD30 antibody conjugated to toxin MMAE, has been studied as monotherapy in r/r cHL patients. Despite high response rates, comprising the overall response rate of 75% and complete responses of 34%, only 9% of all cHL patients achieved long-term remission exceeding 5 years in response to single-agent brentuximab vedotin without any additional therapy.48 Not CD30 downmodulation, but rather MDR1 overexpression has been shown as a putative mechanism of resistance to Brentuximab vedotin in cHL.49 Issues regarding pharmacokinetics, pharmacodynamics of antibody-based approach, and rationale for utilization of another mode of action onto cHL cells have evolved into CD30 CAR-T cells development for cHL.
There are several scFv antibodies commonly used in CD30 CAR-T cell therapy. The most frequently used scFv antibody fragment in CAR-T cell therapy is derived from the mouse IgG1 monoclonal antibody HRS3.50 Clinical trials involving the HRS3 antibody began in the early 1990s, focusing on biodistribution studies in patients with cHL.51 The HRS3 binding module has an affinity of 25 nM. It was successfully humanized for CAR-T cell therapy, proving to be non-inferior in anti-tumor activity compared to the original antibody in the NCG mice model.52,53
Given that murine or even humanized antibodies might trigger a human anti-mouse antibody response, a fully human antibody, 5F11, was produced using a human monoclonal antibody transgenic mouse.54 This antibody demonstrated a much higher affinity of 0.6 nM compared to HRS3 and possessed a different epitope specificity. This is particularly relevant considering that CD30 shedding might reduce the efficacy of CD30 CAR-T cell treatments.55 To address CD30 shedding, the Ki-4 mouse monoclonal antibody developed. Ki-4’s binding to L540 cells prevents the shedding of soluble CD30. This antibody, with an affinity of 2.7 nM, has been explored as part of an immunotoxin but not yet as a binding module of CAR-T.56,57 Another approach to counter soluble CD30 is by targeting a proximal epitope within the CD30 molecule.58 This was investigated with HSP-CAR30, which utilized scFv from the T105 mouse monoclonal antibody.56 This antibody has an affinity of 4.2 nM and has a distinct epitope specificity compared to the aforementioned antibodies.
Currently, the most promising CAR-T targeting modules are nanobodies or VHHs. Due to their small size, they exhibit low tonic signaling in CAR-T, have minimal immunogenicity, and can bind to epitopes that are inaccessible to regular antibodies.59,60 Advances in protein engineering and high-throughput screening have enabled the rapid production of humanized nanobodies for a wide range of targets from diverse synthetic libraries.61 However, CD30 nanobodies and their humanized derivatives remain understudied and should be a focal point in the future of CD30 CAR-T cell therapies.
CD19 is a 95 kDa type I transmembrane protein belonging to the immunoglobulin superfamily and is also a part of the B cell receptor complex.62 CD19 expression initiates during the pre-B lymphocyte stage and persists until plasma cell differentiation, but it is not found on the surface of hematopoietic stem cells.63 CD20 is a 33–37 kDa non-glycosylated tetra-transmembrane protein of the MS4A family proteins. Its expression begins on late pre-B cells and persists until the plasmablast stage of differentiation.64
In a study by Jones et al a small clonotypic population of CD20+CD27+lambda+ALDH+ B cells was identified in cHL cell lines KM-H2 and L428. These cells were capable of differentiating into HRS cells in vitro and maintaining a population of cHL cell lines. The researchers further examined the peripheral blood and affected lymph nodes in 31 patients with cHL, where in 26 of them they were able to identify a monoclonal population of CD19+CD27+ALDH+ cells with restricted immunoglobulin light chains. The median number of such cells was 0.2% (range 0%–2.4%).65 This fact serves as an evidence of existence of a clonotypic population of B cells corresponding to the lymphoma-initiating cells of cHL.
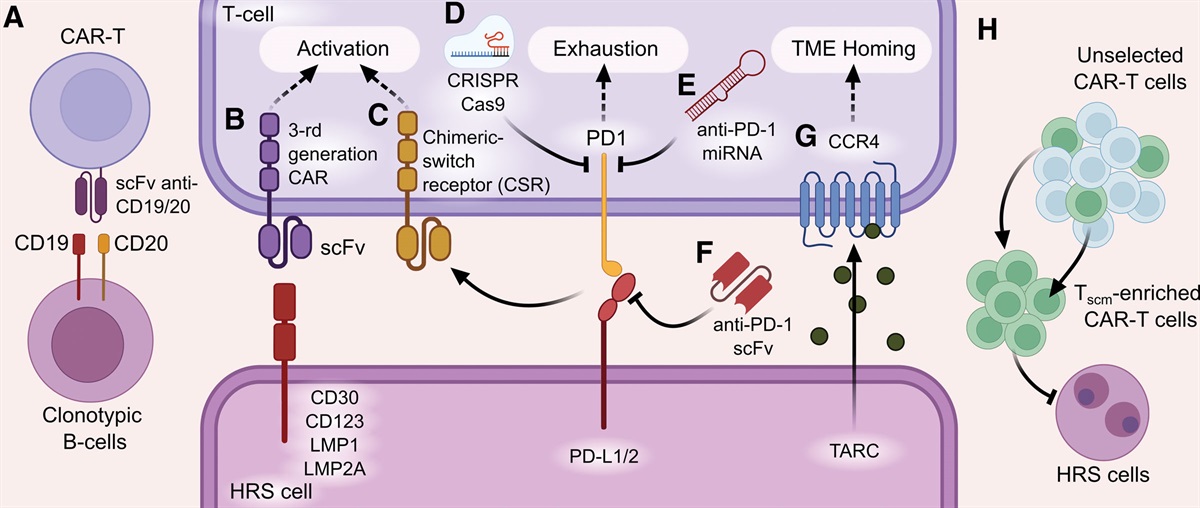
CD20 is expressed on HRS cells in 22% of cHL cases with the median number of CD20+ HRS cells being 55%.66 Moreover, reactive non-tumor B cells could account for >50% of all TME cells. Studies have demonstrated the role of non-tumor B cells in inhibiting the immune response through the production of various immunosuppressive cytokines such as IL-10.67 They also provide proliferative and anti-apoptotic signals through binding ligands of the TNF superfamily (CD30L, CD40L, OX40L) to the corresponding receptors on the surface of HRS cells.68 In a pilot study, 24 patients with r/r cHL were treated with the anti-CD20 monoclonal antibody Rituximab 375/mg twice a week. One patient achieved a complete response and 4 patients achieved partial responses with a median duration of response of 7.8 months. Interestingly, patients with an objective response after treatment with rituximab, according to IHC, did not express CD20 antigen on HRS cells.69 The aforementioned data imply the rationale for the evaluation of CD19/20 CAR-T cells in combination with CD30 CAR-T cells aiming for the eradication of CD19+/CD20+ clonotypic lymphoma cells (Figure 1A) as well as immunosuppressive TME cells (Figure 2A).
 Figure 1.: Strategies for development of CAR-T cells targeting cHL cells. (A) Targeting clonotypic B cells with anti-CD19/CD20 specific CAR-T cells. (B) Targeting HRS cells with anti-CD30, CD123, LMP1, and LMP2A CAR-T cells. (C) Chimeric switch receptor that recognizes ligands such as PDL1/2, LAG3, CD200R, and soluble factors or cytokines (IL-4, TGFb) providing activation signaling.27,70 (D) PD1 gene knockout by CRISPR/Cas9 system or (E) miRNA expression to suppress the translation of PD1 mRNA. (F) Combination of anti-PD1 antibodies with CAR-T cells or production of anti-PD1 antibodies by CAR-T cells themselves. (G) Expression of the CCR4 chemokine receptor for tumor homing along the TARC chemokine concentration gradient. (H) Enriched CAR-Tscm cell product. concentration gradient. (H) Enriched CAR-Tscm cell product. CAR-T cells = chimeric antigen receptor T cells; HRS = Hodgkin and Reed-Sternberg; TME = tumor microenvironment.
Figure 1.: Strategies for development of CAR-T cells targeting cHL cells. (A) Targeting clonotypic B cells with anti-CD19/CD20 specific CAR-T cells. (B) Targeting HRS cells with anti-CD30, CD123, LMP1, and LMP2A CAR-T cells. (C) Chimeric switch receptor that recognizes ligands such as PDL1/2, LAG3, CD200R, and soluble factors or cytokines (IL-4, TGFb) providing activation signaling.27,70 (D) PD1 gene knockout by CRISPR/Cas9 system or (E) miRNA expression to suppress the translation of PD1 mRNA. (F) Combination of anti-PD1 antibodies with CAR-T cells or production of anti-PD1 antibodies by CAR-T cells themselves. (G) Expression of the CCR4 chemokine receptor for tumor homing along the TARC chemokine concentration gradient. (H) Enriched CAR-Tscm cell product. concentration gradient. (H) Enriched CAR-Tscm cell product. CAR-T cells = chimeric antigen receptor T cells; HRS = Hodgkin and Reed-Sternberg; TME = tumor microenvironment. Figure 2.:
Figure 2.: Strategies for development of CAR-T cells targeting TME of cHL. (A) Eradication of normal TME B cells producing IL-10 by anti-CD19 or anti-CD20 CAR-T cells. (B) and (C) Anti-CD123 CAR-T cells, targeting M2 macrophages, mast cells, and eosinophils in the TME. CAR-T cells = chimeric antigen receptor T cell; cHL = classical Hodgkin lymphoma; HRS = Hodgkin and Reed-Sternberg; TME = tumor microenvironment.
CD123, also known as the alpha chain of IL3R (interleukin-3 receptor), is a transmembrane protein with 3 extracellular domains, a transmembrane domain, and a short intracellular domain. The binding of IL-3 to IL3R leads to heterodimerization of α (CD123) and βc (CD131) chains, as well as to the conduction of proliferative and anti-apoptotic signals.71 CD123 is expressed on HRS cells in >90% of cHL cases, with the strongest expression in the nodular sclerosis variant.72 Moreover, CD123 is highly expressed on TME cells, including M2 macrophages, eosinophils, basophils, mast cells, and MDSCs.73
LMP1 is a transmembrane protein of the EBV and carries out signaling similar to CD40, in contrast to which it is constantly active and ligand-independent.74 Six transmembrane domains of LMP1 protein oligomerize and activate its 3 C-terminal cytoplasmic domains, resulting in sustained activation of the NF-κB, MAPK, PI3K pathways, as well as overexpression of the anti-apoptotic protein bcl2.75 These effects result in the conduction of proliferative signals, inhibition of apoptosis and promotion of tumor transformation.76 LMP1 is expressed in 68% of cHL cases.77
LMP2A is an EBV transmembrane protein whose main function is to mimic the missing intracellular signaling of the B cell receptor in HRS cells. LMP2A activates RAS/PI3K/AKT and mTOR pathways, leading to inhibition of HRS cells apoptosis in the germinal center.78,79 LMP2A is expressed in 50% of cHL cases.80 While EBV-specific T cell adoptive cell therapy has been actively studied for the treatment of cHL,81 to our knowledge, CAR-T cells targeting LMP1/2A have not been described in the context of cHL so far. In 1 study, the human antibody that recognizes the extramembrane domain of LMP1 has been developed.82 Later, first-generation CAR incorporating the anti-LMP1 scFv, IgG1 CH2CH3 spacer, CD28 transmembrane, and CD3z cytoplasmic domain has been developed. LMP1 CAR-T cells showed specific killing of LMP1-positive nasopharyngeal carcinoma cell lines and IFNy/IL-2 production in vitro. Most importantly, these cells were able to control LMP1+ tumor growth in an in vivo model.83,84 Further research is needed to assess the activity of LMP1/2A-specific CAR-T cells in cHL models.
PRECLINICAL STUDIES OF CAR-T CELLS DESIGN IMPROVEMENTSBasically, CAR is a synthetic receptor that includes extracellular, transmembrane, and intracellular domains.85 Extracellular domain ensures antigen binding through the ligation by its antigen-recognition domain.8 It could be represented by a single-chain fragment variant composed of heavy and variable light chain regions of monoclonal antibody or other scaffolds like nanobodies,86 ankyrin repeat proteins,87 etc. Antigen-recognition domain is attached to the transmembrane domain by spacer (hinge) responsible for the transmission of receptor-binding signals and anchoring in the host cell membrane. It is usually derived from IgG, CD28 or CD8 molecules. Transmembrane domain plays an essential role in ensuring receptor stability and surface expression.88 After antigen binding an intracellular domain clusters and undergoes conformational changes, leading to the recruitment and phosphorylation of downstream signaling proteins. The intracellular domain of CARs possess an activation domain and 1 or 2 co-stimulatory domains.89 The composition of intracellular domain classifies CARs into 5 generations. The first generation utilizes only one activation domain, most commonly a cytoplasmic domain of CD3 zeta (CD3ζ or z). The second-generation CARs have CD3ζ and 1 co-stimulatory domain, obtained from cytoplasmic domains of co-stimulatory molecules such as 4-1BB (BB), CD28 (28), OX40, CD27, or DAP10. The third generation has 1 CD3ζ and multiple co-stimulatory domains.89 The fourth-generation CARs are engineered to release transgenic different cytokines in the TME.90 The fifth-generation CAR contains a JAK-STAT signaling domain in addition to CD3ζ and 1 co-stimulatory domain.91
Preclinical evaluation of CD30 CAR-T cellsThe first elaboration of first-generation CD30 CAR-T cells began in the 1990s.92 Later, second generation CAR studies and designs taking into account the peculiarities of cHL pathogenesis began to appear. In 1 study, investigators from Baylor College of Medicine have developed CD30-z CAR-T cells, where the starting material of EBV-specific cytotoxic T lymphocytes (CTL) has been exploited. The rationale basis for this lied in the enhancement of persistence of such CAR-T cells by ligation of its cognate TCRs to EBV-infected epithelial, B cells, and antigen-presenting cells as well as targeting HRS cells in EBV-positive cHL cases. Allogeneic and autologous EBV-CTL cells have been transduced with CAR-encoding retroviral vector. After confirmation of stable CAR expression, scientists were able to show a selective killing of CD30+ cHL cell lines (HDLM-2, L428, L540, and KM-H2) as well as EBV-positive lymphoblastoid cell line (LCL) through the CAR and cognate EBV specific TCR. Nor addition of excess CD30L, nor expression of CD30 on the surface of CD30 CAR-T cells did not impair the activity of the CAR-T cells. To demonstrate the functional activity in vivo, CD30CAR EBV-CTLs were transduced with the eGFP-FFluc vector and injected intravenously in sublethally irradiated mice bearing subcutaneous autologous LCL tumor. Homing and expansion at tumor site as well as in vivo activity were monitored by in vivo imaging. Expansion of CAR+ EBV-CTLs at the tumor site of EBV HLA-mismatched LCL tumors was significantly reduced compared to HLA-matched ones. Antitumor activity was also highly dependent on the native EBV-specific TCR receptor as well.93
In the preclinical study, Zhang et al compared second-generation (28z) and third generation (28BBz) CD30 CAR-T cell designs. Although in vitro efficacy was comparable, the third-generation CD30 CAR-T cells exhibited superior effectiveness in an immunodeficient NPG mice model in vivo. Cells of the L428 tumor line were injected into the tail vein of immunodeficient mice, and CD30 CAR-T cells were injected 3 days later. The results indicated that 28BBz significantly increased the lifespan of mice, 50% of which remained alive by day 120 of observation. Moreover, greater persistence (including within tumor tissue), increased production of IFN-γ and reduced exhaustion of CAR-T 28BBz cells have been noted.94
In preclinical studies of HSP-CAR30, investigators from Hospital de la Santa Creu y Sant Pau, Barcelona, developed the third-generation CD30 CAR-T cells.95 The authors have obtained a new CD30 antibody that specifically targets CD30 epitope proximal to the cell membrane, outside the cleavage zone of metalloproteinases. This prevents CD30 CAR-T cells from being inhibited by soluble CD30. Another key aspect of the authors’ approach is an enrichment of Tscm in the final CAR-T cell product. After validating CAR-T cells in vitro, in vivo studies were performed in immunodeficient mice using the L428 intravenous infusion and L540 subcutaneous engraftment models. Infusion of CAR-T cells produced a complete response in all mice in both in vivo models. Following repeated infusion of L428 tumor cells, the tumor development was not observed in mice. This phenomenon was explained by the long-term persistence of CD30 CAR-T cells in the bone marrow and lymph nodes.95
It is known that HRS cells produce chemokines such as CCL17/TARC and CCL22, which are responsible for the homing of Th2 CD4 cells and Tregs expressing the CCR4 chemokine receptor. In the study of Di Stasi et al, it was found that CD8+ T cells weakly express CCR4 on their surface even after activation, which hinders their migration into the tumor tissue. Considering this, the authors developed CD30-28z CAR-T cells that additionally expressed the CCR4 chemokine receptor at the level of 60 ± 19% of CAR-T cells. This led to increased migration along the concentration gradient of the TARC chemokine present in the supernatant of the HDLM-2 and L-428 kLH tumor lines. This pattern of migration was then reproduced in vivo. Immunodeficient mice were subcutaneously inoculated with the tumor cell line Karpas with and without expression of the TARC chemokine. After tumor engraftment, mice were infused with CD30 CAR-T cells with or without CCR4 expression. Starting from day 9, the first group of mice exhibited the accumulation of T cells expressing the CCR4 chemokine receptor in the tumor, while T cells without CCR4 did not migrate into the tumor. The in vivo anti-tumor activity of CD30.CCR4 CAR-T cells was significantly higher than that of CD30 CAR-T cells.96
Preclinical evaluation of anti-CD123 CAR-T cellsInvestigators at the University of Pennsylvania developed CD123 CAR-T cells (CART123) and demonstrated their specific killing ability, proliferation, and production of the cytokines IFNy, IL-2, and TNFa when co-cultured with the HDLM-2, L-428, KM-H2, and SUP-HD1 cHL tumor cell lines. The ability of CART123 to overcome the immunosuppressive effect of M2 macrophages was also demonstrated. CART123 exhibited the simultaneous killing of M2 macrophages and HDLM-2 tumor cells, while M2 macrophages suppressed the activity of CD19 CAR-T cells against the acute B-lymphoblastic leukemia cell line. In another in vivo mice model utilizing HDLM-2 cells, CART123 were injected on day 42, when the tumor mass increased by 20-fold. Within 14 days after CART123 administration, all mice had a complete anti-tumor response, which persisted in 100% of mice for 1 year. The median survival was 128 days in mice treated with mock T cells. Mice without a relapse at day 250 after CART123 were re-injected with HDLM-2 cells. Tumor growth was not observed in this group of mice, and it was accompanied by the re-expansion of CART123 in the blood. Conversely, all mice in the control group showed the development of a tumor.73
CLINICAL TRIALS OF CAR-T THERAPY FOR cHL Second-generation CD30 CAR-T cells incorporating 4-1BB co-stimulatory domainTo date, CD30 CAR-T cells are the most prevalent CAR-T cells for cHL being tested in clinical settings. In Chinese PLA General Hospital, a phase I clinical trial (NCT02259556) is being conducted and has already recruited 17 patients with r/r cHL, 4 (24%) of whom had previously received brentuximab vedotin. Patients received 1 of 3 regimens of lymphodepletion (fludarabine + cyclophosphamide, gemcitabine + cyclophosphamide + chlormethine, or nab-paclitaxel + cyclophosphamide) followed by administration of a CD30-BBz CAR-T cell product at a median dose 1.56 × 107 cell/kg. Adverse events above grade 3 were observed in only 1 patient with cHL (left ventricular systolic dysfunction), which was attributed to previous therapy rather than CAR-T therapy. Non-hematological toxicity associated with CAR-T therapy was manifested by nausea and vomiting (27.8%), urticaria (11.1%), respiratory failure (5.6%), and dizziness (5.6%). In peripheral blood, the peak of expansion of CD30 CAR-T cells occurred between days 3 and 9. At the same time, a higher concentration of the CAR-T cell transgene was determined in the tumor tissue compared to the peripheral blood, indicating their homing into the tumor tissue. Six of 17 (35%) patients achieved partial responses and 6/17 (35%) experienced a stabilization of the disease.97
In the phase I/II clinical trial, HSP-CAR30 (NCT04653649) reported by Caballero et al included 11 patients with r/r cHL or CD30+ T cell non-Hodgkin’s lymphoma. The median age of patients was 49.9 years, and the median number of prior therapies was 4.6. Following leukapheresis, the first 3 patients received 3 × 106/kg of CAR-T cells, 3 additional patients received 5 × 106/kg, and the remaining 4 patients received 10 × 106 cells/kg of CAR-T cells. Lymphodepletion for patients with cHL included a combination of bendamustine and fludarabine. Memory T cells accounted for 93.07 ± 4.8% in the CD4+ T cell compartment and 91.64 ± 4.9% in the CD8+ T cell compartment. Peak concentrations of HSP-CAR30 T cells were observed on average 29 days after infusion and persistence reached 11 months as assessed by flow cytometry. No neurotoxicity was reported in the included patients. Sixty percent of patients developed cytokine release syndrome (CRS) grade 1 and 40% of patients developed a skin rash. Four infectious complications were registered: pulmonary tuberculosis grade 4, CMV pneumonia grade 3, COVID-19, and rhinovirus infection grade 1. Grade 3/4 hematological adverse events presenting as anemia in 50%, thrombocytopenia in 30%, neutropenia in 80%, and prolonged cytopenia in 20% have been reported. 62% of patients with cHL achieved a complete response during the follow-up and a 6-month PFS was 75%.58
Second-generation CD30 CAR-T cells incorporating CD28 co-stimulatory domainPhase I clinical trial (NCT01316146) investigated the safety and efficacy of anti-CD30-CD28z CAR-T cell therapy in patients with r/r cHL and anaplastic large cell lymphoma. The study included 7 patients with r/r cHL, who did not undergo lymphodepletion. Among the 5 patients who received a dose level 3 CAR-T cells (2 × 108 cells/m2), 3 achieved complete responses, which correlated with the highest level of expansion and persistence. Repeated infusions of CAR-T cells were allowed, after which, however, a substantial level of expansion in the peripheral blood was not observed in any patient. A reverse correlation was also established between the level of soluble CD30 on day 0 and the level of subsequent expansion of CD30 CAR-T cells. Notably, in patients who responded to therapy, the increase in the level of soluble CD30 coincided with the peak of the expansion of CD30 CAR-T cells.98
A parallel phase I/II clinical trial (NCT02690545, NCT02917083) enrolled 41 patients with r/r cHL to investigate the safety and efficacy of autologous CD30-28z CAR-T cells. The patients enrolled in this study had a median of 7 prior lines of therapy. Ninety percent of patients had previously received brentuximab vedotin and 81% had received anti-PD1 monoclonal antibody therapy. The study protocol allowed the use of bridge chemotherapy. Lymphodepletion was achieved by bendamustine monotherapy, bendamustine + fludarabine, and fludarabine + cyclophosphamide. CAR-T cell doses administered were categorized into 3 levels: 2 × 107, 1 × 108, or 2 × 108 cells/m2. Repeated infusion was allowed when the stabilization or a partial response was achieved after the first infusion. CRS developed in 10 patients (24%) all of which corresponded to the grade 1. Neurotoxicity was not registered. Thrombocytopenia and neutropenia of grade 3/4, which did not resolve by day 28, developed in 4 patients (10%). Twenty patients (48%) developed a maculopapular rash, predominantly in patients treated with cyclophosphamide-containing lymphodepletion (82%). The overall response rate was 62%, and in the subgroup of patients (n = 32) who received lymphodepletion with fludarabine, the response rate was 72%. Among them, 19 patients (59%) have achieved a complete response, 4 (13%) have a partial response, and 3 patients (9%) have achieved a disease stabilization. The 1-year PFS was 41% in patients who received a fludarabine-containing lymphodepletion regimen and 61% in patients who achieved a complete response. The median PFS was 444 days in patients (n = 19) who have had an active tumor at the time of lymphodepletion or infusion of CAR-T cells and have achieved a complete response. The dose of infused CAR-T cells directly correlated with the peak of expansion, although no correlation with efficacy was observed. Notably, a reduction in CCL17/TARC levels was found to correlate with the treatment response.99
In the phase I/II clinical trial (NCT04288726), Quach et al are currently investigating the safety and efficacy of universal allogeneic CD30 CAR-T cells with a CD28 cytosolic domain, obtained from EBV-specific T cells (CD30.CAR EBVSTs). The study has already enrolled 14 patients with r/r cHL with a median age of 36 years and a history of 5 lines of therapy. Lymphodepletion included fludarabine and cyclophosphamide followed by CD30.CAR EBVSTs at a dose of 4 × 107 to 4 × 108 cells. No cases of graft-versus-host disease (GVHD), neurotoxicity or CRS grade 3 or more were detected. The overall and complete response was 79% and 43%, respectively. However, the persistence of CD30.CAR EBVSTs was not detected after 1 week of infusion.100
The phase II clinical trial Chariot (NCT04268706) enrolled 15 patients with r/r cHL where a median number of previous lines of therapy was 3. The average dose of autologous CD30 CAR-T cells was 2.0–2.7 × 108 cells/m2 after fludarabine + bendamustine lymphodepletion regimen. Patients who did not respond to the first infusion of CAR-T cells were allowed to receive an additional one. The peak of expansion of CAR-T cells was observed on the 7th day, with a persistence of >42 days. After a single infusion of CD30 CAR-T cells, the overall response was 73.3% (n = 11), including 60% complete remissions (n = 9). Among patients who underwent repeated administration of CD30 CAR-T cells (n = 5), the overall and complete response rates were 100% and 60%, respectively. Neurotoxicity and CRS grade 3/4 have not been reported.101
Third-generation CD30 CAR-T cells incorporating 4-1BB and CD28 co-stimulatory domainsIn the phase I study conducted at Tongji Medical College (ChiCTR-OPN16009069), 6 patients with cHL and 3 patients with anaplastic large cell lymphoma were enrolled. Patients received lymphodepletion with a combination of fludarabine and cyclophosphamide followed by infusion of third-generation CD30-28BBz CAR-T cells at a median dose of 1.4 × 107 cells/kg. Six (66%) patients developed CRS, 4 of which were grade 1 or 2. No neurotoxicity was reported. Most patients had persistent lentiviral copies for up to 6 months. Among the 6 patients with cHL, 5 achieved complete responses, which were durable for up to 38 months. Five patients received antiPD1 consolidation therapy 90 days after CAR-T cell therapy and one upon the disease relapse with subsequent disease stabilization.102
New design CAR-T cells and anti-PD-1 combination studies in cHLGrover et al are conducting a phase I/II clinical trial, in which patients with r/r cHL and CD30+ T cell lymphoma of the skin are being treated with autologous CD30 CAR-T cells expressing the chemokine receptor CCR4 (CCR4.CD30.CAR-Ts). The study has already enrolled 12 patients, 10 of whom had r/r cHL. Following the lymphodepletion regimen with bendamustine and fludarabine, patients were infused with CCR4.CD30.CAR-Ts at doses ranging from 2 × 107 to 1 × 108 cells/m2. All patients had previously received brentuximab vedotin, 11 patients received anti-PD1 therapy, 9 patients received auto-HSCT and 5 patients received allo-HSCT. The median number of previous treatment regimens was 5.5. One patient developed grade 1 neurotoxicity, and no grade 3 or higher of CRS have been observed. The efficacy of therapy was evaluated in 8 patients with cHL: 6 patients (75%) achieved a complete remission and 2 patients (25%) achieved a partial remission. At a median follow-up of 12.7 months, median PFS and OS have not been reached.103
Mei et al have registered an Ib/II Action clinical trial to investigate the safety and efficacy of autologous CD30 CAR-T cells in combination with nivolumab. The study protocol will include cHL patients r/r to the first-line therapy. Following leukapheresis, patients should undergo 2 cycles of nivolumab 480 mg once every 4 weeks, followed by lymphodepletion with the inclusion of fludarabine and bendamustine. Subsequently, patients will receive an infusion of autologous CD30 CAR-T cells at a dose of 2 × 108 cells/m2 followed by 2 additional cycles of nivolumab with the evaluation of efficacy after the treatment. After that, one group of patients will undergo auto-HSCT and the second group will receive maintenance therapy with nivolumab.104
Ramos et al have observed a CD30 expression retainment in relapsing tumors after CD30 CAR-T cell therapy, suggesting that recurrence was attributable to insufficient persistence of CAR-Ts within the highly immunosuppressive TME of cHL.99 The first case of decreased expression of CD30 on the surface of HRS cells after brentuximab vedotin and CD30 CAR-T cell therapy in a patient with cHL has been described by Kim et al.105 Later, Marques-Piubelli et al.106 compared CD30 expression levels before and after CD30 CAR-T cell therapy in 4 patients with cHL and found a decrease in expression levels according to IHC in all cases.
CAR-T cells targeting CD19 antigen in cHLThe other potential explanation for the lack of long-term responses after CD30 CAR-T cell therapy in cHL is the maintenance of a pool of clonotypic CD19+ cHL cells as well as presence of immunosuppressive B lymphocytes in the TME. Investigators from the University of Pennsylvania have conducted a phase I/II clinical trial to investigate the safety and efficacy of CD19 CAR-T cell therapy in patients with r/r cHL (NCT02277522, NCT02624258). The second-generation CAR receptor was utilized, incorporating 4-1bb and CD3z domains in the cytoplasmic region. The delivery of the transgene was achieved through mRNA electroporation into autologous lymphocytes. Lymphodepletion was performed with cyclophosphamide at a dose of 30 mg/kg for 4 days −4 to −1 day and on day 7. CD19 CAR-T cells were infused on days 0, 2, 4, 9, 11, and 14. The dose of the cell product was dependent on body weight. Patients weighing <80 kg received 8 × 105 to 1.5 × 106 CAR-T cells/kg and patients with body weight over 80 kg—1 × 108 CAR-T cells/kg. No non-hematological toxicity has been registered. Among the 4 patients infused with mRNA CD19 CAR-T cells, one achieved a complete response, one had a partial response, one experienced stabilization, and one did not achieve an objective response. Notably, patients with longer persistence of CAR-T cells demonstrated more pronounced responses.107 These findings demonstrate that clinical responses in cHL could be achieved by the elimination of CD19+ HRS cells, clonotypic B cells (Figure 1A), and TME B cells (Figure 2A) by CD19 CAR-T cells. These results highlight the potential of CD19 CAR-T cells as a promising approach in the management of cHL. Nevertheless, one should anticipate already described resistance mechanisms to CD19 and CD20 CAR-T cell therapy comprising the loss of antigen expression as a result of mutation-selection or alternative splicing, as well as transcriptional, post-transcriptional, and post-translational events.64,108–110 Further research is needed to elucidate the efficacy and resistance mechanisms of CD19/CD20 CAR-T cells in the context of cHL.
Considering the entirety of clinical data on CAR-T cell therapy in cHL, initial findings suggest the potential for improved CAR-T cell designs. Strategies such as increasing the dose of CAR-T cells, implementing lymphodepletion with bendamustine and fludarabine, conducting CCR4 chemokine receptor transduction, and enriching the CAR-T cell final product with Tscm (Figure 2G, H) are being explored to enhance treatment outcomes. However, larger studies are necessary to validate these trends. A comparative summary of the aforementioned clinical studies is provided in Table 1.
Table 1 - Clinical Trials Results of CAR-T Cell Therapy for cHL Design of CAR-T cells Phase Patients, n Platform LD Dose Outcomes CRS/ICANS References CD30-BBz I
留言 (0)