Human serum albumin (HSA) constitutes more than 60 % (by mass) of proteins in human blood plasma [1]. It is a monomeric, globular protein with turns and extended loops that is predominantly α-helical [2], [3], [4]. The circulatory system contains HSA, which is the most abundant carrier protein in human serum [5]. It is highly water-soluble, with blood concentrations ranging from 30 to 50 g/L (∼0.53–0.75 mM) and a pH range of 7.35–7.45 [6] HSA has several ligand binding sites, which is consistent with its exceptional ability to bind a diverse spectrum of ions and compounds [7], [8], [9]. As a result, HSA has emerged as a leading candidate as a molecular cargo and nano-vehicle in the biophysical, medicinal, and industrial applications [10], [11], [7]. As reported in 1992, X-ray crystallography was used to determine the three-dimensional structure of the first HSA (PDB ID: 1UOR) [12]. Since then, several structures in apo and bound-ligand forms have been found, mostly via single crystal X-ray crystallography, and deposited in the Protein Data Bank (PDB). HSA, in its mature and active state, has 17 disulfide bridges forming the protein fold with one free cysteine at position 34 (CYS34) [13].
HSA (66.5 kDa) is a helical and heart-shaped protein made up of three domains that are similar in topology and structure: domain I (residues 1–195), domain II (residues 196–383), and domain III (residues 384–585) [14] (See Fig. 1). In each domain, there is one antiparallel six-helix (subdomain A) and four-helix (subdomain B) motif [4]. There are two main flexible ligand binding sites located in subdomains IIA and IIIA [15], with a strong affinity for various molecules on HSA called Sudlow's sites (I and II), which is in accordance with X-ray resolved HSA structures and supported by several biophysical analyses [16], [17]. Furthermore, seven fatty acid (FA) binding sites, each with distinct affinities for FAs [18], have been reported, and two of them overlap Sudlow's sites. Primarily, physiological ligands of HSA are considered to be FAs. A total of nine long-chain FAs with varying affinities [19], [3] and asymmetrically arranged across HSA can be bound at distinct binding sites (i.e., FA1–FA7). There are two main categories of FA binding sites: high affinity (FA2, FA4, FA5) and low affinity (FA1, FA3, FA6, FA7) [20].
HSA is a major carrier of drugs targeting multiple organs and tissues because of its high blood concentration and multi-binding characteristics [23], [24]. Nearly 2.5 percent of drugs (234 approved, 12 experimental, 3 investigational, 3 withdrawn) are reported to interact with HSA, according to DrugBank version 5.1.1 issued on July 3, 2018 [25], [26], [27], [28]. Many drugs' pharmacokinetic behavior is influenced by HSA-drug binding, which alters their efficacy and delivery rate [7], [29]. As a result, obtaining a detailed understanding of HSA-ligand binding and interactions can be extremely beneficial in the development and design of novel drugs [30], [31]. Predicting the binding of investigational drugs to HSA, as a way of discribing possible drug-ligand interactions, drug-drug interactions and drug bioavailability; this has been a subject of research by various scientists [15], [22]. Co-administration of drugs may lead to competition for the same HSA binding sites which can significantly enhance the free fraction of the low-affinity drug. Also, there could be competition between drugs and endogenous substances for certain HSA binding sites, thus posing potential clinical consequences [32], [33], [34]. Most of the predictive protocols explored in the past for albumin binding had some challenges, including high flexibility of binding site conformations after docking and poor resolution of crystallographic HSA structures in the protein data bank. In this study, we endeavored to base our data collection on various analyses from crystallographic structures with high-resolution power [15], [22], embarking on a completely different approach from docking and molecular dynamic (MD) simulations. Herein, we provide an alternative yet robust predictive model for ligand binding to HSA binding sites using carefully curated and experimentally validated data. Various chemoinformatic, statistical, and computer-aided drug discovery (CADD) algorithms were employed in a deductive workflow.

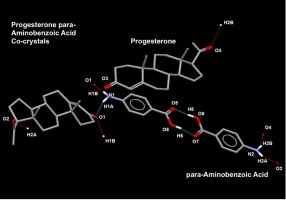
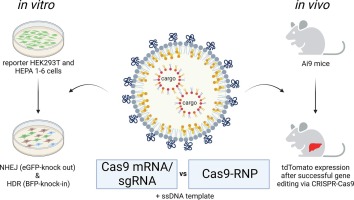
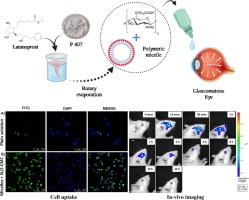
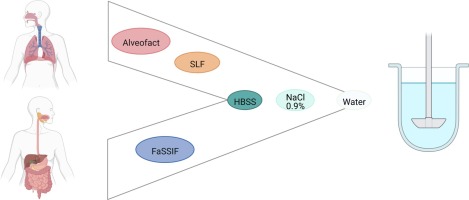

留言 (0)