
記住我
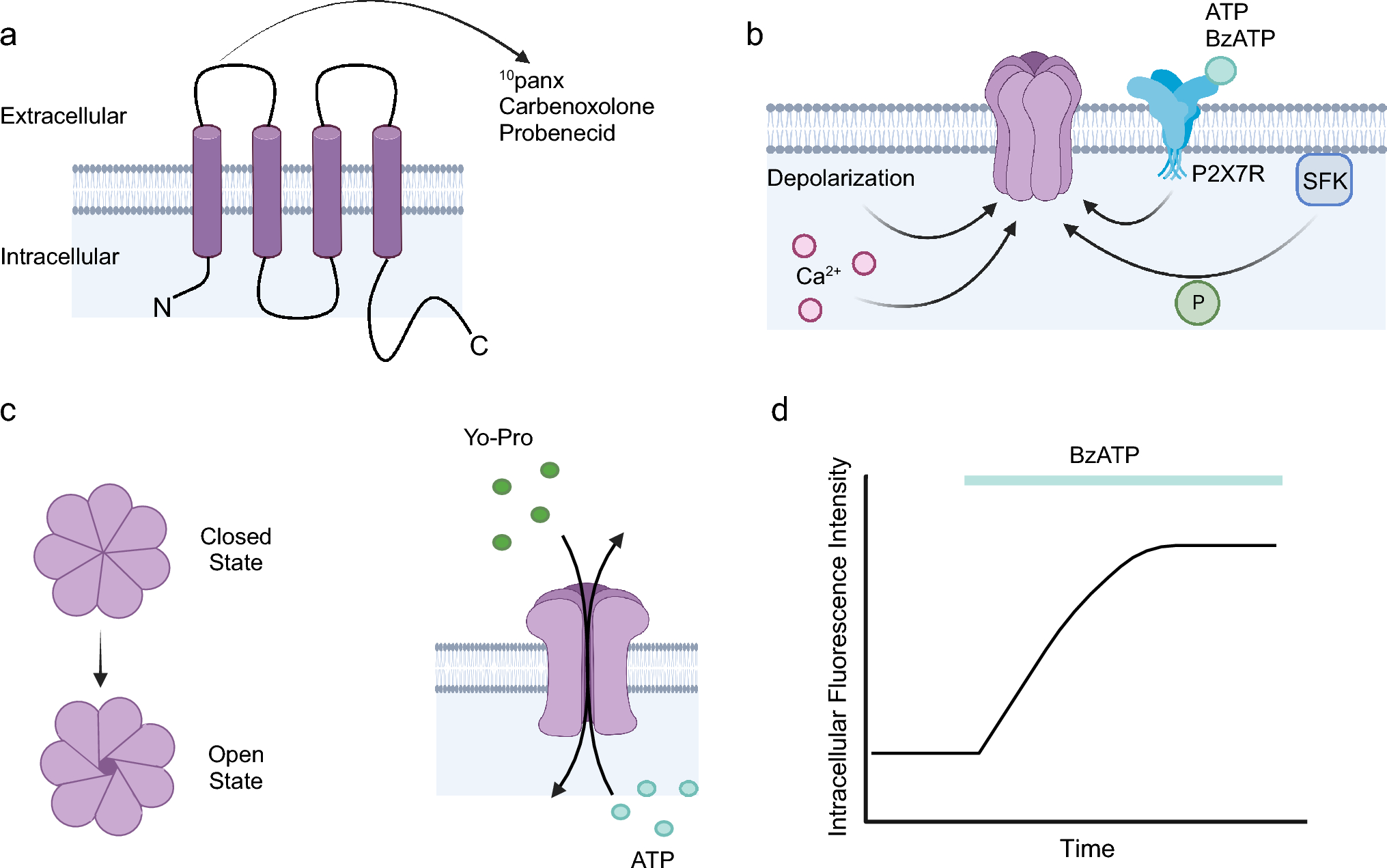

For the SAD part, eligible volunteers were enrolled in a single-blind, partially randomised, placebo-controlled parallel group study. Initially, volunteers were allocated to receive BI 1358894 3, 6, 10, 25, 50, 100, and 200 mg under fasted conditions, or BI 1358894 200 mg under fed conditions. The volunteers were then randomised within each dose group to receive BI 1358894 or placebo in a 3:1 (drug:placebo) randomisation ratio. Each dose group comprised six volunteers receiving BI 1358894 and two volunteers receiving placebo. Prior to receiving study treatment, volunteers were fasted, or, for the BI 1358894 200 mg fed group, received a high-fat, high-calorie breakfast. Each dose was investigated sequentially in ascending order, with each subsequent dose group only being investigated following a dose escalation review. Plasma PK samples were collected following BI 1358894 administration as follows: 3 mg dose group, Days 1–5; 6–200 mg (fasted) dose groups, Days 1–9; 200 mg (fed) dose group, Days 1–7 and then on Days 9, 11, 15, 22, and 29 [Fig. 1a]. For the quantification of BI 1358894 plasma concentrations in the blood samples, 2.7 mL of blood was drawn from the forearm vein into a potassium ethylenediaminetetraacetic acid anticoagulant blood drawing tube, tightly capped and stored at −20°C. The plasma samples were extracted by protein precipitation in a 96-well plate. An aliquot (50 μL) was mixed with 250 μL of an internal standard solution (10 nmol/L BI 1358894) dissolved in acetonitrile/methanol/water (50/45/5, v/v/v), centrifuged, and 160 μL supernatant was mixed with 200 μL of 10 mM ammonium formate buffer (pH 4). Blank human plasma was used for the preparation of calibration samples, quality control samples and blank samples. Chromatography was performed using an analytical reversed-phase ultra performance liquid chromatography column (ACQUITY UPLC BEH Shield RP18) with 10 μL of samples. BI 1358894 concentrations in plasma were analysed by a tandem mass spectrometer with electrospray ionisation in the positive ion mode.
Fig. 1
Design for study 1402-0001 for (a) the SAD part and (b) the food effect part. *Plasma samples for PK assessment of BI 1358894 in the 3 mg fasted dose group were collected on Days 1–5 following BI 1358894 administration. In the BI 1358894 6–200 mg dose groups, plasma samples for PK assessment were collected on Days 1–9 following BI 1358894 administration. In the BI 1358894 200 mg fed group, plasma samples for PK assessment were collected on Days 1–7, and then on Days 9, 11, 15, 22 and 29, following drug administration. A allocation, D day, PK pharmacokinetic, QD once daily, S screening, SAD single ascending dose
2.1.2 1402-0001 Study Part 2For the food effect part, eligible volunteers were enrolled in an open-label, randomised, two-way crossover study. Volunteers were randomised to two treatment sequences: fed followed by fasted, or fasted followed by fed. For each sequence, they received either BI 1358894 50 or 100 mg as a single dose after fasting or after having received a high-fat, high-calorie breakfast. There was a washout period of at least 7 days between treatment administrations within each sequence. Plasma PK samples were collected on Days 1–9 following BI 1358894 administration (Fig. 1b) and analysed as reported in Sect. 2.1.1.
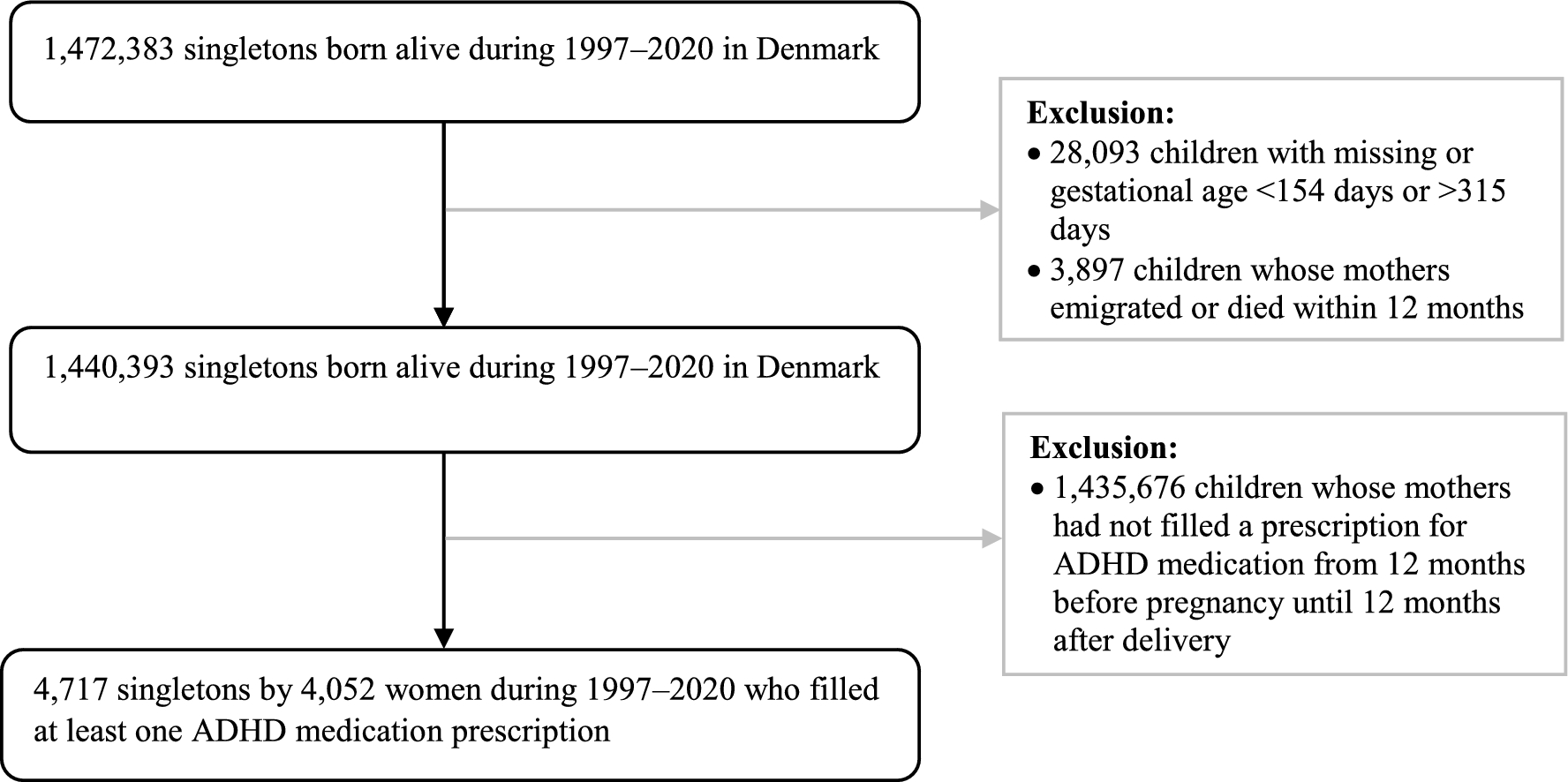
2.1.3 1402-0002 Study Part 1For the MAD part, eligible volunteers were initially allocated to a BI 1358894 dose group (10, 25, 50, 100, or 200 mg) before being randomised to receive BI 1358894 or placebo (4:1 ratio) within each group. Volunteers received BI 1358894 or placebo once daily for 14 days under fed conditions. Each dose of BI 1358894 was investigated sequentially, in ascending order, with each subsequent dose group only being investigated following a dose-escalation review. Plasma samples for PK assessment of BI 1358894 were collected on Days 1–7, Day 9, Day 11, and Days 13–22 (Fig. 2) and analysed as reported in Sect. 2.1.1.
Fig. 2
Study design for study 1402-0002. A allocation, D day, MDZ midazolam, PK pharmacokinetic, QD once daily, S screening
2.1.4 1402-0002 Study Part 2The DDI assessment was conducted by using a microdose of midazolam as part of the regular MAD trial (part 1). Midazolam (75 µg) was administered to volunteers across all dose groups 1 day prior to receiving their first dose of BI 1358894 or placebo, and in parallel to receiving BI 1358894 or placebo on Days 1 and 14. It should be noted that the DDI PK data have been published elsewhere and will not be reported here [12, 13].
2.2 ParticipantsBoth phase I studies were conducted in healthy male volunteers at sites in Germany. Demographic data are presented in Tables 1 and 2. Volunteers were included in the studies if they were healthy according to the investigator’s assessment, were 18–45 years of age, and had a body mass index (BMI) of 18.5–29.9 kg/m2. Volunteers were excluded if they had any finding in the medical examination deviating from normal, laboratory values outside the reference range, or evidence of concomitant disease considered clinically relevant by the investigator. Other exclusion criteria included the use of drugs that might influence the study results within 30 days prior to administration of study treatment, smoking (>10 cigarettes per day), a previous history of suicidal ideation, or alcohol or drug abuse. Recruitment was limited to male volunteers as conclusive data on reproductive toxicology were not yet available for these early clinical studies.
Table 1 Participant demographics and baseline characteristics by treatment group for the single ascending dose part of study 1402-0001Table 2 Participant demographics and baseline characteristics by treatment group for the food effect part of study 1402-00012.3 Endpoints and Assessments2.3.1 1402-0001The primary endpoint for study 1402-0001 was the number of volunteers with drug-related adverse events (DRAEs). Serious adverse events (SAEs) were defined as any AE that resulted in death, was immediately life-threatening, required inpatient hospitalisation, prolonged existing hospitalisation, resulted in persistent disability, was a congenital anomaly/birth defect, or was deemed serious for any other reason. Cancers of new histology and exacerbations of existing cancer were classified as an SAE regardless of the duration between discontinuation of the drug. An adverse event of special interest (AESI) was defined as any specific AE identified as being of particular concern for prospective safety monitoring and assessment within the trial; hepatic injury was the only protocol-specified AESI. The intensity of AEs was classified as mild (awareness of signs/symptoms that were easily tolerated), moderate (enough discomfort to cause interference with usual activity) or severe (incapacitating or causing inability to work or perform usual activities).
Safety was further assessed by collating treatment-emergent AEs (TEAEs), routine laboratory tests, and the evaluation of vital signs, electrocardiograms (ECGs), visual analogue scales (VAS), and suicidality assessment via the Columbia Suicidal Severity Rating Scale (C-SSRS).
The secondary PK endpoints of interest were the area under the concentration–time curve (AUC) of BI 1358894 in plasma from administration time (t = 0) to the last quantifiable data point (AUC0–tz); AUC of BI 1358894 in plasma from administration time (t = 0) extrapolated to infinity (AUC0–∞); and maximum measured concentration of BI 1358894 in plasma (Cmax).
2.3.2 1402-0002For study 1402-0002, the primary endpoint was the number of volunteers with DRAEs. The SAEs and AESIs were as defined in Sect. 2.3.1. Safety was also evaluated using TEAEs, laboratory tests, vital signs, ECGs, VAS, and the C-SSRS.
The secondary PK endpoints of interest were AUC of BI 1358894 in plasma from administration time (t = 0) to 24 h (AUC0–24) after administration of the first dose; Cmax of BI 1358894 in plasma after administration of the first dose; AUC of BI 1358894 in plasma at steady state over a uniform dosing interval (AUCτ,ss) after the last dose; and Cmax of BI 1358894 in plasma at steady state over a uniform dosing interval (Cmax,ss) after the last dose.
2.4 Ethical ConsiderationsThe studies were carried out in accordance with the principles of the Declaration of Helsinki, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Good Clinical Practice (GCP) guidelines, applicable regulatory requirements and Boehringer Ingelheim standard operating procedures. All participants provided informed written consent in accordance with ICH GCP and local procedures. The study protocol was reviewed and approved by the local independent ethics committees and relevant local authorities.
2.5 Statistical AnalysisFor both studies, all primary safety and PK endpoints were calculated descriptively. For study 1402-0001, the PK parameters of AUC and Cmax were assessed for dose proportionality using a regression model applied to log-transformed data. Based on the estimate for the slope parameter, a two-sided 95% confidence interval (CI) of the slope was computed. The relative bioavailability based on AUC0–tz, AUC0–∞ and Cmax was analysed using an analysis of variance (ANOVA) model on log-transformed data. Exposure ratio of test versus reference treatment (geometric mean [gMean]) was calculated.
The SAD part planned to include 64 volunteers; this was not based on a power calculation. The size of eight volunteers per dose group (six receiving BI 1358894 and two receiving placebo) is commonly used in SAD studies and is considered sufficient for the exploratory evaluation of single-dose safety and PK endpoints. For the food effect part, a maximum of 24 volunteers (12 receiving BI 1358894 50 mg and 12 receiving BI 1358894 100 mg) were planned for enrolment. With this sample size, a certain precision in estimating the ratio of gMeans could be expected with 95% probability. As this was a first-in-man trial, no information on intrasubject variability was available.
For study 1402-0002, dose proportionality was assessed using a linear regression model applied to log-transformed data. Based on the estimate for the slope parameter, a two-sided 90% CI for the slope was computed. Attainment of steady state was analysed for each dose level by a repeated measures linear model on a logarithmic scale using the trough predose concentrations of the analyte in plasma immediately before administration of the Nth dose after N-1 doses were administered (Cpre,N). Trough concentrations of Cpre,9, Cpre,11, Cpre,13 and Cpre,14, and the concentrations taken directly at the end of the first and last dosing interval of BI 1358894, were used.
The MAD part planned to include 50 volunteers, which was not based on a power calculation. The size of 10 volunteers per dose group (eight receiving active treatment and two receiving placebo) is commonly used in MAD studies and is considered sufficient for the exploratory evaluation of multiple-dose safety and PK endpoints.
留言 (0)